
Theoretical and Practical Aspects of Alloy Plating - The 16th William Blum Lecture - Part 1
This article is the first of three parts of a re-publication of the 16th William Blum Lecture, presented at the 62nd AES Annual Convention in Toronto, Ontario, Canada on June 23, 1975. Dr. Ernst Raub presents a comprehensive treatise on alloy plating.



by
Ernst Raub



Forschungsinstitut für Edelmetalle und Metallchemie
Schwäbisch-Gmünd, Germany
Recipient of the 1974 William Blum
AES Scientific Achievement Award
Originally published as Plating & Surface Finishing, 63 (2), 29-37 (1976), this article is the first of three parts of a re-publication of the 16th William Blum Lecture, presented at the 62nd AES Annual Convention in Toronto, Ontario, Canada, on June 23, 1975. A printable PDF version of Part 1 is available by clicking HERE. The printable PDF version of the complete 44-page paper is available HERE
ABSTRACT
The enormous literature on alloy plating is mostly restricted to alloys in the strict sense which contain metallic components with at least some per cent of a second metal. For practical purposes, codeposition of a second metal and also of non-metallic elements, e.g., sulfur or selenium, in small amounts is often interesting. Hydrogen as an alloying element has been known for a long time, but only incompletely investigated. The same is true for the activity of hydrogen ions in the electrolyte. The influence of electrodepositing conditions, e.g., of specific inhibition, cathodic adsorption and incorporation and of the effects of complexing agents present, is discussed. Properties of electrolytic deposits, their changes by heat treatment, and alloy formation between deposit and underlying metal or between two or more electrolytic layers are mentioned.
Introduction and background
William Blum and George B. Hogaboom1 wrote in 1949, in the third Edition of their book Principles of Electroplating and Electroforming, on alloy deposition, "In a strict sense every plating bath of a single metal is an alloy bath, since both metal and hydrogen may be discharged and the hydrogen may dissolve in or form compounds with the metal deposited."
In the same publication the authors mentioned the existence of an enormous amount of literature on alloy deposits. Abner Brenner2 gives an extensive description on the Electrodeposition of Alloys, which comprises two volumes with 1370 pages. Therefore it is a hazardous enterprise to talk about alloy deposition in a short paper. And yet I will try to do so because the electrodeposition of alloys holds problems of general interest for electroplating, which even today are only partially cleared. Furthermore the practical importance of alloy plating has grown during recent years, especially for various new applications in modern industry.
For a long time brass and gold-alloy deposits have been employed as decorative deposits, but for some decades brass plating has also been used for technical purposes to improve bonding of rubber to steel or to enhance the deep drawing properties of steel sheet. Gold-alloy deposits are used in the modern electronic industry as tarnish resistant surface layers with low contact-resistance. Tin-lead deposits are employed to guarantee good solderability of printed circuits in electronics. For the same purpose nickel-tin deposits are applied.
They also find rising interest in other industries. Tin-lead and to a certain degree tin-indium deposits have been used for many years as linings for friction bearings. At present electrodeposited magnetic alloys, which contain as main components iron and nickel or cobalt with different additional elements, serve as memory elements in the electronics industry.3 Electrolytic alloy plates have been used for many years as wear resistant materials. Nickel-cobalt alloys serve for the production of tools by electroforming. Other metal deposits with small amounts of alloying metals are used for bright and / or hard deposits. Some so called hard-bright silver layers contain antimony. Gold alloys with gold contents as low as possible while still yielding sufficient chemical resistance, are not only deposited in all colors for decorative purposes but also for industrial use to minimize the use of the expensive gold. For the same reason the deposition of palladium alloys, such as palladium-nickel and palladium-copper, find rising interest.
Generally in practice deposits with small amounts of a second metal or of non-metallic elements are no longer considered to be alloys, but they often have such significant properties that it is impossible to neglect them.
It may be mentioned that electrolytically produced compound materials, to which dispersion hardened deposits also belong, are only mixtures of metals and non-metallic substances with quite different properties, and therefore will not be discussed here.
Of special relevance to alloy deposits in the strict sense is the molecular inclusion of non-metallic, organic or inorganic substances. Not only is their influence on the properties of the deposited metal similar to that of a codeposited second metal, but quite often alloy deposition is connected with molecular inclusion. Therefore it is necessary to discuss it with alloy deposition.
The formation of alloys by diffusion between deposit and basis metal or double and triple layers is of practical interest. Occasionally an annealing of alloy deposits is necessary to promote diffusion and to achieve certain properties. Another class of alloy deposits, formed by autocatalytic chemical reduction without an external current source, will not be discussed in this paper.
Since up to now electrolytic deposition of alloys has been carried on for the most part in aqueous solutions, this report will be restricted to the cathodic reactions in aqueous solutions, and the properties of the deposits dependent upon the deposition conditions, including the influence of subsequent heat treatment.
During the passage of current the cathode reacts with all the substances contained in the electrolyte. Besides cathodic reduction, chemical reactions in the electrolyte and physical adsorption on the cathode surface are of importance. Often the adsorbed substances do not exist in the original electrolyte. They are formed by cathodic reduction and subsequent chemical reactions. Cathodic adsorption and inhibited crystallization were investigated by H. Fischer.4-7 For electrodeposition of metals and alloys partial coating by physical adsorption is most important. Chemisorption or chemical reactions with the cathode surface may cause surface layers which exhibit an unfavorable influence.
The cathodic reduction reactions not only include metals and hydrogen, but also involve non-metallic elements which are present as oxides, or as sulfur, selenium or phosphorous compounds. They may be cathodically reduced to negatively charged ions, such as S+4 to S-2 ions. Since the solubility of heavy metal sulfides is small, cathodic adsorption and inclusion takes place readily. Sulfonic acids and other organic sulfur compounds used as addition agents also undergo cathodic reduction and sulfide formation. Adsorption and inclusion of non-metallics as elements or insoluble compounds, e.g., heavy metal sulfides, do not necessarily presume a cathodic reduction of oxidic compounds. They take place too when soluble compounds present as addition agents chemically decompose in the electrolyte, e.g., by hydrolysis, during formation of elements or negative ions.
Cathodic reduction is not limited to simple cations, but also extends to neutral molecules and anions.
Metals which exist in several valences may be reduced under proper conditions by cathodic reduction to a lower valence without metal deposition. When the bath contains a component which forms an insoluble compound with a metal ion of lower valency, this may be of decisive influence on the deposition of alloys.
For the discharge of ions, their migration in the electric field is not critical as is the transport of the dischargeable mass particles to the cathode by diffusion and convection. At sufficiently high concentrations of reducible mass particles in the cathodic diffusion layer, their reduction potentials are the controlling factor for the reactions actually taking place on the cathode surface. Concerning the discharge of metal ions one has to keep in mind that in most practical cases electrolytes are used which contain the metals to be discharged, as complex ions, quite often as anions. Normally in these electrolytes the discharge step does not result from the predominant complex, but from a lower coordinated complex, or from a totally different compound. Our knowledge of the mechanism of discharge from complex cyano ions is due largely to the work of H. Gerischer.8,9 The discharge of zinc in cyanide electrolytes proceeds via Zn(OH)2 and not via the Zn(CN)4-2 complex predominant in the electrolyte. In the cyanide cadmium bath the discharge of the cadmium goes via Cd(CN)3ˉ or Cd(CN)2, dependent on the cyanide concentration of the bath as the discharge controlling step. In fact, especially in cyanide electrolytes mass particles which exist only in small amounts control the cathodic discharge. They are formed by chemical reactions of the predominant complexes. So it is possible to influence the discharge potential of a metal in an extremely wide range by changing the concentration of complexing agents in the electrolyte. The stability of the predominant complex ion in the cyanide solutions of many metals can be changed to a great extent, but differing for every metal, by merely varying the cyanide concentration of the electrolyte. The stability of the predominant complex controls the formation of the lower coordinated dischargeable complex, and thus the discharge potential of the metals. It is possible to achieve similar discharge potentials of metals, or to reverse potentials, by altering the concentration of the complexing agent.10 In cyanide brass-plating, the zinc content of the deposits increases with increasing cyanide content of the electrolyte because the stability of the copper cyanide complex Cu(CN)3-2 or even Cu(CN)4-3 is much stronger at higher cyanide concentration. The discharge of copper in the cyanide bath is controlled by the Cu(CN)2ˉ complex. Its formation depends on the stability of the predominant higher coordinated complex.
Addition of sodium hydroxide to the electrolyte favors codeposition of zinc because it shifts the equilibrium,8
Zn(CN)4-2 + 4OHˉ ⇔ Zn(OH)4-2 + 4CNˉ
to the right, and facilitates the formation of the dischargeable Zn(OH)2. Many other examples of the influence of the complexing agent concentration on alloy plating can be given, especially when several complexing agents are present.
The total quantity of the complexing agents and their ratio to the metal in the electrolyte may be much more decisive for the composition of the deposited alloys than the concentration ratio of the metals to be deposited. This is valid as long the limiting current density of one of the metals is not exceeded.
Furthermore, due to the formation of complexes with low solubility, the discharge of the more noble metal may be inhibited by cathodic adsorption, so that the codeposition of the less noble metal is facilitated.
During electrodeposition a steady state occurs at the cathode, which is controlled by several factors, e.g., the ratio of the dischargeable or reducible mass particles in the cathodic double layer, their average concentration in the electrolyte, the convection and diffusion processes, the number of mass particles reduced per unit of time, and the replacement of substances included during electrodeposition.
As a rule of thumb it may be stated that under normal conditions the discharge of the most noble metal is favored, causing deposits to be richer in this metal than could be expected from its relative concentration in the electrolyte. All changes of electrolysis conditions which increase polarization, such as rising cathodic current density, decreasing temperature, unstirred electrolyte, do shift the composition of the deposit in direction to the less noble metal. All changes of the depositing conditions which diminish the polarization, such as decreasing current density, rising temperature and stirred electrolyte, increase the content of the more noble metal in the alloy. This rule is only applicable as long as the limiting current density is not surpassed. Above the limiting current density the cathodic discharge is diffusion-controlled since the concentration of dischargeable particles at the cathode surface is zero. As soon as the limiting current density for the deposition of one metal is obtained any further increase of the current density has no additional influence on the electrodeposition of this metal. If the current density is higher than the limiting current density for both metals then the ratio of the metals in the deposit will not change with increasing current density. Any current rise will only produce a third cathodic reduction reaction, e.g., hydrogen evolution.
Molecular inclusion of chemical compounds
The influence of the inclusion of non-metallic substances has been investigated long ago, and has been thoroughly discussed by A. Brenner.2 In spite of this, I want to mention the main points. The molecular inclusion of non-metallic substances may cause changes of properties which are more pronounced than those resulting from the codeposition of an alloying metal. Indeed, by inclusion the real properties of the alloy may be masked completely.
Such changes of properties are only proportional to the included substances in the lower range of inclusion. At high inclusion levels, no clear relationship upon the quantity of the included substance exists.
By molecular inclusion the hardness of copper can reach about 300 kp/mm2, of silver nearly 200 kp/mm2 and of iron up to 800 kp/mm2.
Hard iron layers attain the hardness of chromium deposits, and their wear resistance equals that of chromium.
Table 1 shows the abrasion resistance of deposits with different hardnesses.
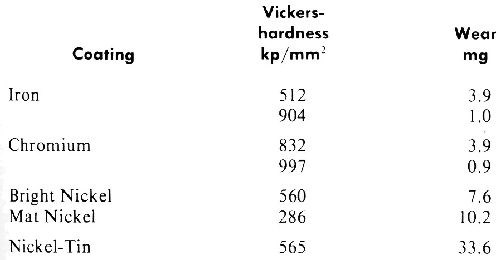
Table 1 - Wearing with emery powder of different electrolytic coatings (load, 50 g; path length, 1000 m).
As seen from the table, under the abrasive conditions used, the harder chromium and iron deposits investigated had reduced wear. At corresponding hardnesses of both metals, the abrasion resistance is nearly the same. However, the decrease of abrasion with increasing hardness is not a general rule. Quite often high hardness resulting from inclusion is associated with high embrittlement, causing a decrease in abrasion resistance as can be seen from Table 1 for tin-nickel alloys.
Higher inclusion rates influence the electrical resistance in a very pronounced way. Due to the formation of isolating films or filaments in the electrocrystallized metal, the resistivity may be higher by three orders of magnitude than that of the pure metal. Also the chemical properties change significantly.11
Heat treatment of deposits with included non-metallic substances effects characteristic changes of their properties as shown for the electrical resistivity of silver in Fig. 1. At a certain relatively low temperature (the decomposition temperature of the non-metallics) resistivity drops suddenly.11
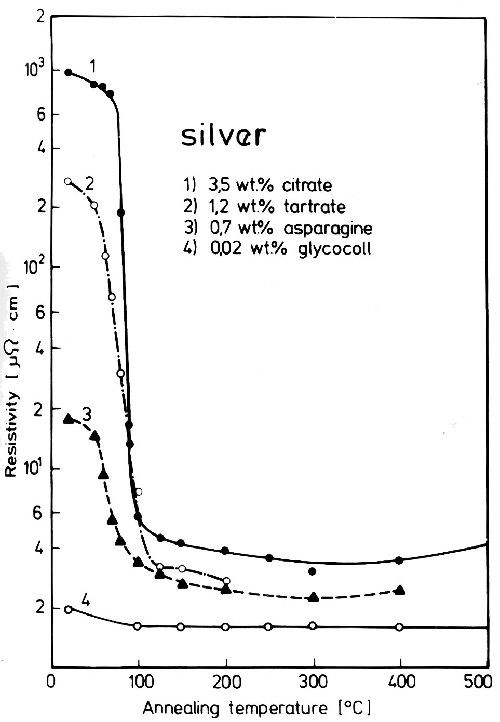
Figure 1 - Resistivity changes of silver deposits with included non-metallics during heat treatment.
Figure 2 shows the influence of non-metallic inclusions on the hardness of iron deposits and its change by heat treatment at different temperatures. The low hardness of pure iron deposits changes only very little by heat treatment. In the presence of more inclusions hardness reaches higher values. But no clearcut relationship with the amount of included substance is known. Again, during heat treatment hardness quickly decreases at a distinct temperature. In Fig. 2 the hardness curve for the iron deposit with 95.5 wt% iron shows a peak at 700°C. This iron sample contained citrate, which decomposed with the segregation of carbon. At temperatures of about 700°C the incorporation of this carbon again increases hardness.12
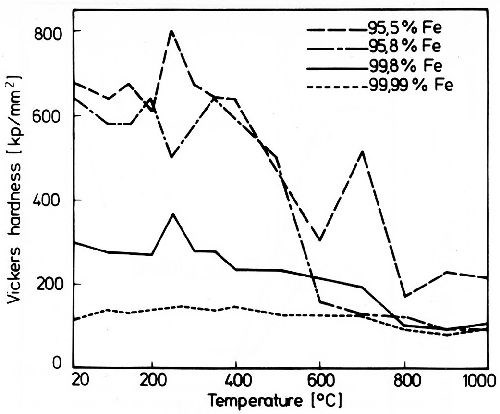
Figure 2 - Hardness changes of iron deposits during heat treatment.
Deposits with included substances have a lamellar structure, which may be coarse or very fine. Partially they show, by x-ray investigations, a characteristic fibre texture but they may also be completely disordered. The interference lines of the Debye-Scherrer diagrams are indistinct and may completely disappear at high diffraction angles. With high quantities of incorporated material the x-ray structure may approach that of the amorphous state. The classic example is the so called explosive antimony. According to x-ray studies the molecular inclusion of non-metallic substances in the metal lattice causes an extremely high amount of lattice defects, and finally only very small coherent lattice ranges exist.
Concerning their properties, electrodeposited pure metals are comparable to hard worked metal if the deposition temperature is much lower than the temperature at which lattice defects heal quickly. Electrodeposited metals with incorporated non-metallic substances are comparable to extremely age hardened supersaturated solid solutions. A difference exists insofar as, in age hardened alloys, by diffusion and agglomeration at a longer heat treatment, the normal properties of the alloy are regained. The influence of non-metallic inclusions in the lattice of an electrodeposited metal cannot be lost in this way as non-metallic compounds do not diffuse in the metal lattice. It is lost only by decomposition of the included material. The decomposition is mostly combined with gas evolution, so that the remaining metal will show gas porosity, and therefore the properties of the annealed pure metal are not completely equaled (see Fig. 1).
An age hardening effect of metal deposits with only low non-metallic inclusion, as shown in Fig. 3 for gold,13 is caused by the greater thermal expansion of the included non-metallic substance compared to that of the metal before its decomposition. The same age hardening effect was also observed during tempering of other metals with only very low inclusion, e.g., silver.14
The close relationship between alloy and single metal deposits with inclusion of non-metallic substances explains why under certain circumstances it is not possible to examine the influence of the alloying metal separately from that of an inclusion. It can only be discerned when incorporation remains small, and no coherent films or filaments of included substances exist. Under such conditions it is possible to investigate those properties which are only slightly structure sensitive, such as lattice constant and electrical resistance, which are controlled by the ratio of the metallic components in the electrodeposited alloys.
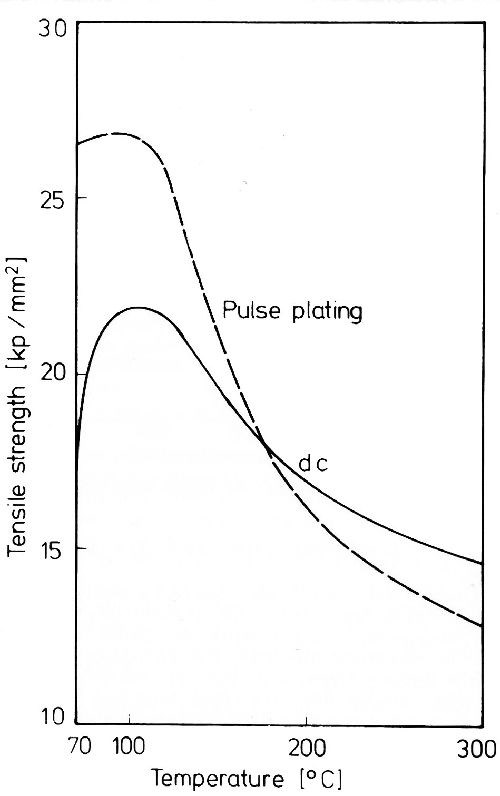
Figure 3 - Tensile strength changes of gold deposits during aging; bath composition: 10 g/L KAu(CN)2, 30 g/L KH2PO4, 30 g/L K2HPO4, pH 6, 70°C, 0.3 A/dm2, pulse plating and DC.
Hydrogen and alloy deposition, hydrolysis and abnormal alloy deposition
The co-discharge of hydrogen is in many aspects of great importance in the deposition of alloys.
In acid solutions hydrogen is discharged via protons. In alkaline electrolytes the proton concentration is very small, and the discharge of hydrogen occurs immediately by decomposition of water molecules. The earlier assumption that the first step is a discharge of alkali metal ions is now considered to be wrong.
Deposition of quite a number of metals is only possible because of the high hydrogen overvoltage on their surface. The importance of hydrogen for the deposition of alloys of metals such as tungsten and molybdenum, together with iron group metals, which cannot be deposited in the unalloyed state from aqueous solutions, was thoroughly discussed by A. Brenner.2 The generally known discharge of sodium ions on mercury cathodes is not only due to the high hydrogen overvoltage, but is also due to the formation of a diluted sodium-mercury alloy, which equals the electrochemical properties of mercury. The hydrogen ion activity of the electrolyte is not only of interest for the amount of hydrogen codeposition, but often determines the properties of the alloy-electrolyte. In weak acid solutions the pH will rise in the cathodic diffusion layer by hydrogen discharge, and hydrolysis of metal salts may occur. The hydroxide compounds formed in this way are adsorbed on the cathode surface and influence the electrodeposition of alloys.
During metal deposition under conditions which do not lead to significant cathodic hydrogen evolution hydrolysis may occur by changes in the average pH of the electrolyte. Normally, by hydrolysis, the cathodic potential curve will be shifted to higher overvoltages. This is shown in the upper part of Fig. 4 for cadmium, a metal which electrocrystallizes from sulfate solutions with very low overvoltage. At pH values between 1 and 6, the overvoltage of cadmium changes very little. But at a pH of 7, at which hydrolysis and adsorption of Cd(OH)2 on the cathode surface take place, the overvoltage is significantly higher.15
Zinc sulfate solutions show hydrolysis and cathodic adsorption of hydrolysis products at rather low pH values. The influence of pH on the overvoltage of zinc is much stronger and quite different from what it is for cadmium. As seen from the lower part of Fig. 4 for zinc, rising pH lowers the overvoltage. This points out that the discharge or zinc via Zn(OH)+ and Zn(OH)2 takes place more readily than via Zn+2.
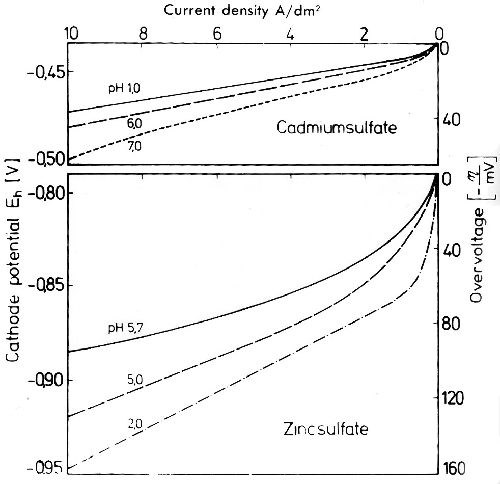
Figure 4 - Potential- and overvoltage-current density curves in 0.5M cadmium- and zinc-sulfate solution at different pH values.
In contrast the deposition of nickel is strongly inhibited by the presence of even small quantities of zinc in the electrolyte, as seen in Fig. 5.16 By additions of zinc to the nickel electrolyte, the cathodic potential shifts to that of zinc. In pure sulfate solutions the deposition potential of zinc is, under comparable conditions, 200 to 300 mV more negative than that of nickel. The striking slope of the potential curves in the alloy baths is due to zinc hydroxide adsorption on the cathode surface.
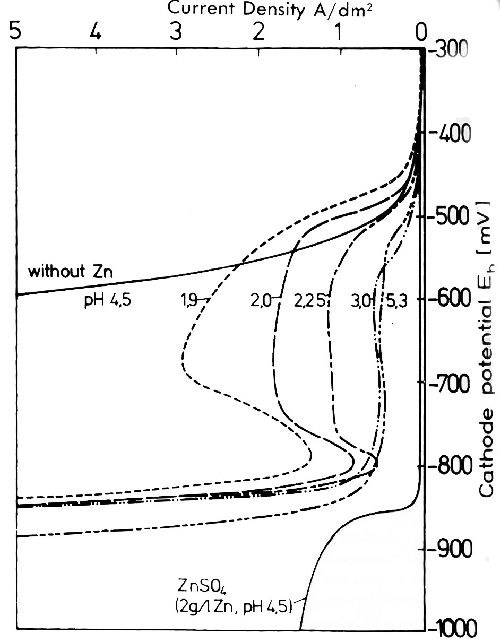
Figure 5 - Influence of 30 m mole/L Zn on the cathode potential current density curves of a Watts nickel bath at different pH values.
In a Watts bath containing zinc the electrochemical behavior of the cathode is just the same as in the presence of organic inhibitors, such as Butindiol, N-Methylphthaleneimid a.o.17 Figure 6 shows curves for the zinc content of nickel-zinc alloys from a Watts bath with small additions of zinc. As can be seen, the zinc to nickel ratio of the deposits at lower current density is much greater than that in the solution. With rising current density it decreases very rapidly as a result of the depletion of zinc in the cathodic diffusion layer.18 The preferred deposition of zinc compared to nickel is remarkable, because in sulfate solutions of the pure metals, nickel is the more noble metal (Fig. 5). But in a bath containing nickel and zinc, zinc assumes the behavior of the more noble metal. This is caused by the strong inhibition of the zinc hydrolysis products on nickel deposition, and their depolarization on zinc deposition. This is an example of the so-called abnormal electrodeposition of alloys which is characterized by preferred deposition of the less noble metal.
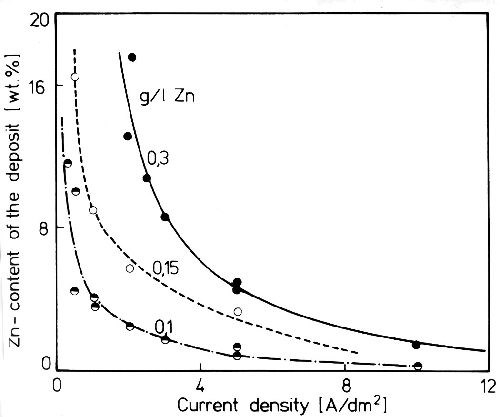
Figure 6 - Zinc content of nickel deposits from a Watts bath with 1 g/L p-toluol sulfonamide and small additions of zinc at 50°C and stirred electrolyte.
Cobalt and iron deposition is also strongly inhibited by zinc hydroxide so that abnormal alloy deposition can be observed.19
The importance of pH for brass plating has been discussed. Other examples are acid gold plating baths which contain gold as Au(CN)2ˉ. The pH value of these baths must be kept in relatively close limits. If the pH is too low the cyanoaurate complex decomposes and gold cyanide segregation begins. If the pH is too high, alloying metals may segregate as insoluble compounds; for instance as ternary phosphates. On the other hand, the stability of the complexes may be raised so much at a higher pH that they are no longer discharged with the gold; e.g., in the presence of EDTA. The pH value is of special importance to the stability of the sulfite gold alloy baths.20
Sulfur and selenium in alloy deposits
Up to the present time solid solutions of sulfur in metals during electroplating have not been conclusively proven. Iron deposits with higher sulfur contents have some lattice changes which indicate a certain solubility of sulfur in electrodeposited iron.21 The influence of sulfur on the properties of the metal deposits is equal to that in melted or sintered alloys of corresponding composition.
In general, sulfate is not cathodically reduced under normal electroplating conditions. In alkaline plating solutions also sulfite cannot be reduced. Figure 7 shows the strong influence of current density on the sulfur content of nickel deposits from electrolytes with additions of sodium hydrogen sulfite (left) and aromatic sodium sulfonic acids (right). In the presence of sodium hydrogen sulfite, the sulfide contents of the deposits are much higher than in the presence of aromatic sulfonates. In the latter the sulfite ion is formed at first by cathodic reduction from the aromatic sulfonate. This sulfite is then reduced to sulfide, and cathodic adsorption and inclusion of nickel sulfide take place.22,23 The inclusion of nickel sulfide is diffusion controlled. Sulfur and sulfide contribute to depolarization in the deposition of nickel. In plating practice aromatic sulfonates and related compounds are generally used as stress reducers in bright nickel baths, a disadvantage being the lower corrosion resistance of these deposits. Potentiostatic anodic potential current density curves in 0.1N sulfuric acid (Fig. 8) show that nickel deposits produced in electrolytes with sulfur-free addition agents have only a very narrow current density range in which active solubility appears. Sulfur-containing nickel deposits are actively dissolved over a wide range of current density, and show no real passivity. Figure 9 shows that between sulfur-free and sulfur-containing nickel in weakly acid sodium chloride solution, a galvanic element is formed in which the sulfur-free nickel is about 200 mV more noble than sulfur-containing nickel. Sulfur-free addition agents have no significant influence on the potential of nickel deposits. Sulfur-containing nickel is more susceptible to contact corrosion than sulfur-free nickel, which must be remembered for practical applications. The poorer corrosion resistance of sulfur-containing nickel is due to the fact that sulfide prevents passivation by formation of nickel sulfide on the corroding surface. Therefore the difference in the corrosion resistance of sulfur-containing and sulfur-free nickel is only existent in corrosion media free from sulfurous acid. In the presence of sulfurous acid, nickel sulfide forms on the surface of sulfur free nickel deposits according to the reaction:
4 SO3-2 ⇔ 3SO4-2 + S-2.
Therefore a difference in corrosion mechanism can no longer be observed.24,25
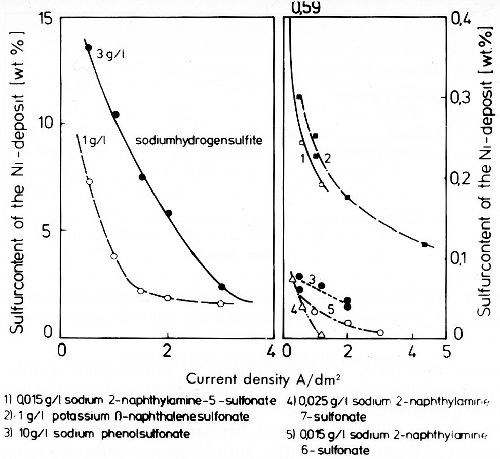
Figure 7 - Sulfur content of nickel deposits from nickel baths with additions of sodium hydrogen sulfite or aromatic sulfur compounds.
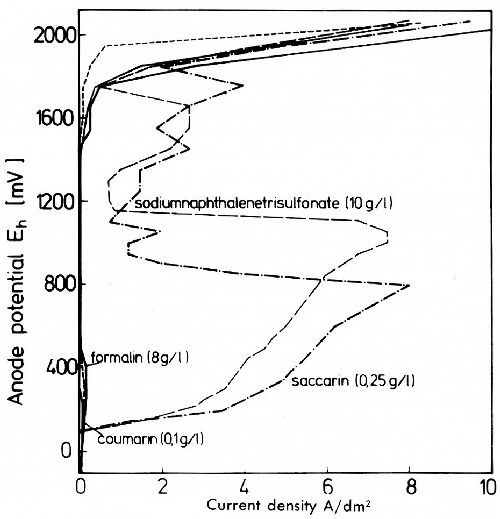
Figure 8 - Potentiostatic anode potential current density curves of electrolytic nickel from a Watts bath with different addition agents.
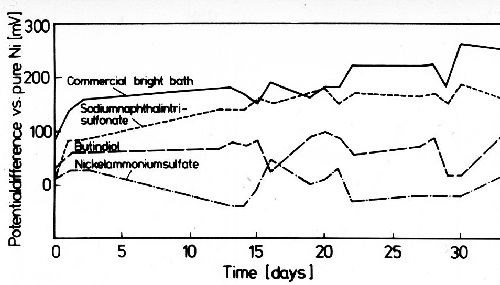
Figure 9 - Potential difference between pure electrolytic nickel and nickel from baths with different addition agents in 3% sodium chloride solution with a pH of 3 at room temperature.
In baths where the sulfite ions are not cathodically reduced, the effect of sulfur or sulfide may be achieved by addition of a soluble sulfur compound. From this a metal sulfide or elementary sulfur may be formed by a chemical reaction. Substances of this kind are thiourea and thiosulfate.26-28
The interesting influence of very small amounts of S-2 ions on silver deposition from the cyanide bath may be discussed here.29 Figure 10 shows potentiostatic cathode potential curves for an electrolyte with 80 g/L Ag and 200 g/L free KCN under nitrogen. As cathode, a silver sheet was used. At as low a concentration as 0.06 ppm S-2 a discontinuous increase in current density is observed between -0.6 and -0.8 V. Above -0.9 V the potential current density curves for the S-2-free and the S-2-containing electrolyte are equal. The influence of the S-2 concentration in the electrolyte is also shown in Fig. 10. This behavior can be used to determine very small S-2 quantities between 2 and 0.01 ppm. The influence of such small sulfur amounts on the cathodic potential curve in cyanide silver solutions is caused by the extremely low solubility of silver sulfide. As a consequence the reaction,
2Ag(CN)2ˉ + S-2 ⇔ Ag2S + 4CNˉ
takes place.
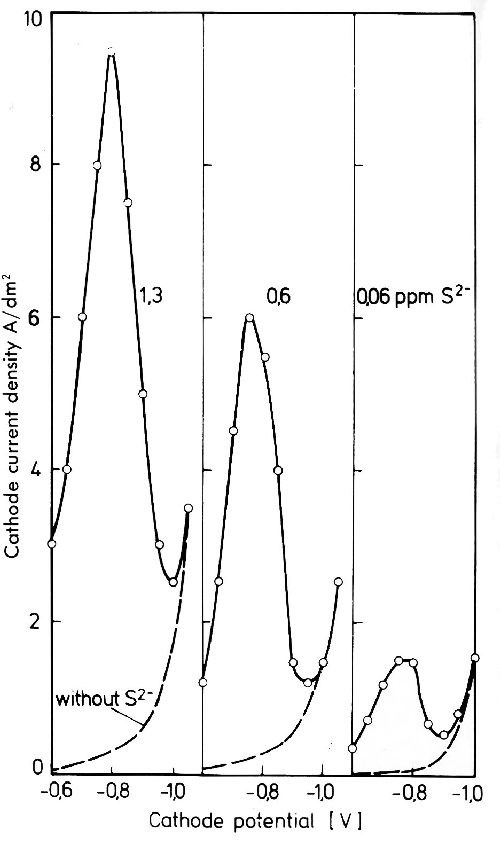
Figure 10 - Potentiostatic potential-current density curves in a cyanide silver electrolyte with and without additions of S-2 ions. Electrolyte composition 200 g/L KCN and 80 g/L Ag as KAg(CN)2.
The silver sulfide is adsorbed on the cathode surface and is included by the crystallizing silver. It depolarizes the discharge of silver. Its influence on the slope of the cathode potential curve lasts until all S-2 present is included as silver sulfide by the crystallizing silver.
To a certain extent, selenium shows an influence similar to sulfur, but the selenate and selenite ions are much more easily reduced than the corresponding sulfur compounds. Selenous-and selenic acids are chemically reduced in acid solutions by most metals under formation of elemental selenium. In general, soluble selenides decompose under segregation of selenium. Cathodic reduction of selenates also occurs in acid solutions. In alkaline electrolytes investigated, cathodic reduction of selenates was observed only in the cyanide copper bath. Selenites are electrolytically reduced in acid and some alkaline baths.30,31
The codeposition of selenium in acid solutions is diffusion controlled.
The selenite ion is simultaneously reduced in the alkaline cyanide gold electrolyte, but not in the cyanide silver bath. Therefore the properties of silver, such as its brightness, are not improved by addition of selenium dioxide. To influence silver deposition, it is necessary to use selenide compounds which chemically decompose in the electrolyte, e.g., selenocyanate or selenourea.
As seen from the pulse rates of gold deposits from a gold bath with additions of doped selenium dioxide (Fig. 11) the codeposition of selenium is small and drops relatively quickly with increasing current density. Over 0.3 A/dm2 it is nearly independent of the current density. When selenium dioxide is used as brightener for gold baths it is to be noted that oxidation of the selenite to the selenate ion on insoluble anodes must be prevented.
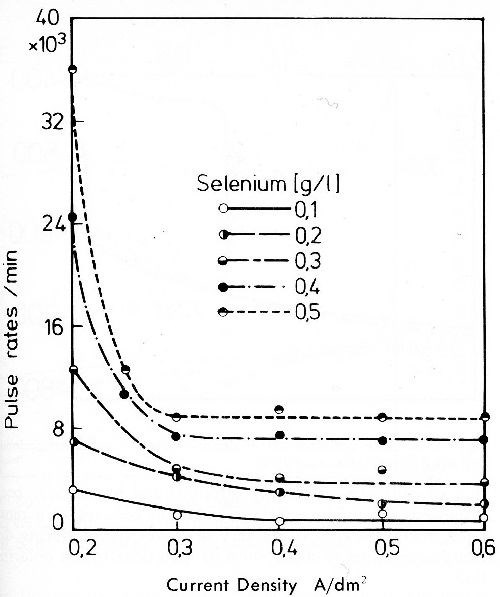
Figure 11 - Pulse rates of gold deposits from a stirred bath with 2 g/L Au as KAu(CN)2 and 4 g/L KCN with additions of sodium selenite doped with75 Se (bath temperature 55°C).
In the chromium bath selenic acid is stable, and is reduced simultaneously with chromium. Selenite in the chromium plating bath is slowly oxidized to selenate. As long as selenous acid is present it is impossible to get reproducible results.
Selenic acid cannot be used in place of sulfuric acid as a catalyst for chromium deposition; it is reduced preferentially with formation of grey amorphous selenium. The codeposition of selenium in the chromium plating solution is controlled by the penetration polarization. From Fig. 12, it can be seen that by codeposition of selenium, macro- and microcracked as well as microporous chromium may be deposited.




Figure 12 - Influence of selenium on the surface structure of chromium deposits (electrolyte: 250 g/L CrO3; 2.5 g/L H2SO4; 44°C.
Alloy deposition from simple cations
In a few cases, deposition potentials are so close together that simultaneous discharge of two or more metals can be achieved from simple ions without difficulties. It is possible for the deposition of iron, nickel and cobalt alloys. The same is true for tin-lead alloys. Figure 13 shows a 50 mV difference between pure tin versus pure lead and only 2 or 3 mV between different alloy compositions.32 Table 2 shows that in alloy plating the deposition of lead is favored over tin. This is in agreement with the slope of the cathodic potential curves in solutions of the single metals. The behavior during the simultaneous discharge of lead and tin in fluoborate solution corresponds to the values of the standard potentials, Pb/Pb+2 = -0.126 V and Sn/Sn+2 = -0.140 V. This is one of the few cases in which the standard potential of two metals permits a prediction of the deposition of alloys. A prerequisite for this is that the controlling step is the discharge of metals from simple, and not from complex ions. Furthermore it is necessary that the deposition polarization in the electrolyte employed is nearly equal for both metals.
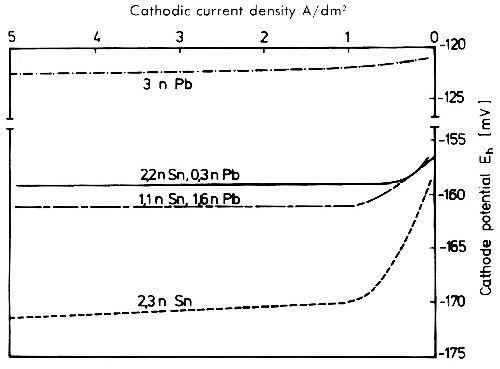
Figure 13 - Cathode potential-current density curves of lead and tin in fluoborate solutions.

Figure 14 - Cathode potential-current density curves in quiescent cadmium and nickel sulfate solutions at 20°C.
In this aspect codeposition of cadmium and nickel is interesting. Standard potential of Ni/Ni+2 is -0.23 V and of Cd/Cd+2 -0.402 V. Therefore one might assume that during simultaneous discharge nickel is favored. This is wrong. As already discussed, cadmium is discharged in sulfate solutions with very low and nickel with very high polarization. This causes cadmium to be discharged in sulfate solutions at a potential 0.2 to 0.3 V more noble than nickel,33 which explains why cadmium behaves more noble than nickel (Fig. 14). The limiting current density of cadmium is reached in an electrolyte with 2.5 g/L Cd at about 0.7 A/dm2. In a nickel electrolyte with 30 g/L Ni and 1.65 g/L Cd, the deposition potential of cadmium turns to more negative values but it is still very significantly more noble than nickel. Figure 15 gives the composition of cadmium-nickel alloy deposits from baths with varying cadmium concentrations as a function of the current density. At low current density (dependent on the cadmium concentration) only cadmium is deposited. At current densities higher than the limiting current density of cadmium, codeposition of nickel begins. This different behavior of cadmium and nickel ions during the cathodic discharge makes possible a quantitative electrolytic separation of the two metals by operating at current densities below the limiting current density of cadmium. The mechanism of the preferred deposition of zinc in zinc-containing nickel baths is quite different from that of cadmium-containing solutions and has already been discussed. In sulfate solutions cadmium behaves as a more noble metal because of its very low overvoltage during discharge compared to nickel with its high penetration polarization.
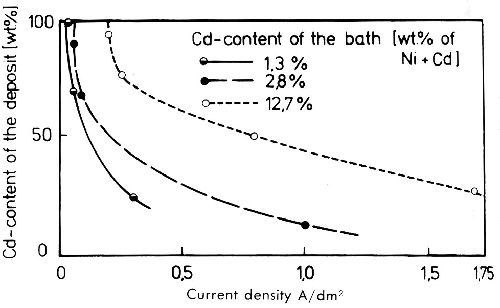
Figure 15 - Cadmium content of cadmium-nickel alloy deposits from sulfate solutions at different cadmium contents of the electrolyte.
Specific inhibition and alloy plating
The inhibition of nickel, cobalt and iron by zinc hydroxide is one case of specific inhibition. In other alloy plating baths specific inhibition may be achieved by addition agents which form a complex of low solubility with the more noble metal, and thereby inhibit its discharge by cathodic adsorption. The behavior of thiourea in acid copper solutions may be discussed here.27,34-37 Thiourea is oxidized by Cu+2 to dithioformamidine with formation of copper(I) ions and hydrogen. Under proper experimental conditions, separation of an insoluble copper(l) thiourea complex can be observed. This complex, a strong inhibitor for copper deposition, is adsorbed on the cathode surface and incorporated. The preceding chemical reduction of copper with thiourea is not just assumed. During passage of current in an acid copper electrolyte, besides metal deposition, small quantities of Cu+ ions are formed. These react immediately on the cathode with thiourea and the formation of the complex. This reaction is of interest, e.g., in the codeposition of lead and copper from acid solutions. In Fig. 16, cathode potential-current density curves in a perchloric acid solution21 are given. The deposition potential of copper is about 0.4 V more noble than that of lead, and in the mixed solution only copper is deposited as long as its limiting current density is not exceeded. But when only 1 g/L thiourea is added to the electrolyte, the potential curve of copper is shifted by about 0.45 V to more negative values, whereas the lead potential remains unchanged. From the thiourea-containing electrolyte, lead is codeposited with copper at the lowest current densities. In this electrolyte, lead still shows the properties of the less noble metal. The lead-to-copper ratio in the deposits is lower than that in the bath. It rises with the current density, a characteristic of less noble metals. To summarize, the influence of thiourea on copper deposition in acid solutions is not that of a normal complexing agent, but of an inhibitor by cathodic adsorption, and therefore its influence can be observed at small concentrations in the electrolyte.
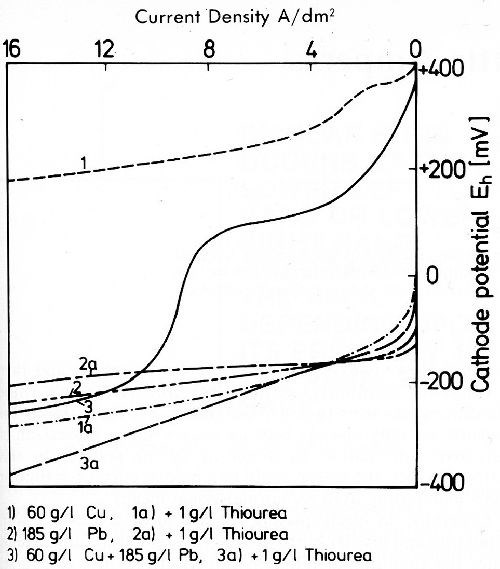
Figure 16 - Cathode potential-current density curves of copper and lead in acid perchlorate solutions with and without thiourea, at 25°C and quiescent electrolyte.















Related Content

A Chromium Plating Overview
An overview of decorative and hard chromium electroplating processes.
Read More
Possibilities From Electroplating 3D Printed Plastic Parts
Adding layers of nickel or copper to 3D printed polymer can impart desired properties such as electrical conductivity, EMI shielding, abrasion resistance and improved strength — approaching and even exceeding 3D printed metal, according to RePliForm.
Read More
How to Choose Between Sulfate and Chloride-Based Trivalent Chromium
There are several factors to consider when choosing between sulfate and chloride-based baths for trivalent chromium plating. Mark Schario of Columbia Chemical discusses the differences and what platers should keep in mind when evaluating options.
Read More
Innovation in Plating on Plastic
Plating on advanced plastics solution offers improved adhesion, temperature resistance and cost savings.
Read MoreRead Next

A ‘Clean’ Agenda Offers Unique Presentations in Chicago
The 2024 Parts Cleaning Conference, co-located with the International Manufacturing Technology Show, includes presentations by several speakers who are new to the conference and topics that have not been covered in past editions of this event.
Read More
Delivering Increased Benefits to Greenhouse Films
Baystar's Borstar technology is helping customers deliver better, more reliable production methods to greenhouse agriculture.
Read More
Episode 45: An Interview with Chandler Mancuso, MacDermid Envio Solutions
Chandler Mancuso, technical director with MacDermid Envio discusses updating your wastewater treatment system and implementing materials recycling solutions to increase efficiencies, control costs and reduce environmental impact.
Read More