
Theoretical and Practical Aspects of Alloy Plating - The 16th William Blum Lecture - Part 2
This article is the second of three parts of a re-publication of the 16th William Blum Lecture, presented at the 62nd AES Annual Convention in Toronto, Ontario, Canada on June 23, 1975. Dr. Ernst Raub presents a comprehensive treatise on alloy plating.



by
Ernst Raub



Forschungsinstitut für Edelmetalle und Metallchemie
Schwäbisch-Gmünd, Germany
Recipient of the 1974 William Blum AES Scientific Achievement Award
Originally published as Plating & Surface Finishing, 63 (3), 30-43 (1976), this article is the second of three parts of a re-publication of the 16th William Blum Lecture, presented at the 62nd AES Annual Convention in Toronto, Ontario, Canada, on June 23, 1975. A printable PDF version of Part 2 is available by clicking HERE. The printable PDF version of the complete 44-page paper is available HERE.
Alloy deposits from complex compounds
As an example, cyanide complexes are widely used in commercial practice to approach deposition potentials of the metals to be simultaneously deposited. The classical example, copper-zinc, has already been mentioned.
Stability of cyanide complexes differs strongly and the complex from which discharge occurs changes with metal-to-cyanide ratio of the electrolyte.
The deposition potentials in cyanide solutions are not always close enough together to permit the codeposition desired. Simultaneous deposition of zinc and cadmium happens equally in acid sulfate and alkaline cyanide solutions.38 In Fig. 17 the potential curves in the acid and in the alkaline cyanide bath are shown. They are nearly 0.4 V apart, and codeposition of zinc with cadmium occurs only when the limiting current density of cadmium is exceeded. In the cyanide alloy bath at higher current densities a second limiting current density is observed which relates to the deposition of zinc. As in the acid bath, in the alkaline cyanide bath also, zinc deposition does not take place until the limiting current density for cadmium is exceeded. The often described simple method to transform a cadmium electrolyte into a zinc electrolyte just by substituting cadmium anodes for zinc anodes relies on the preferred cathodic discharge of cadmium and the anodic dissolution of zinc. Using this method one has to keep in mind that at current densities above the limiting current density for cadmium separate crystallization of both metals easily leads to the deposition of alloys with insufficient properties. It is better to work during the conversion of a cadmium into zinc bath at current densities below the limiting current density of cadmium.
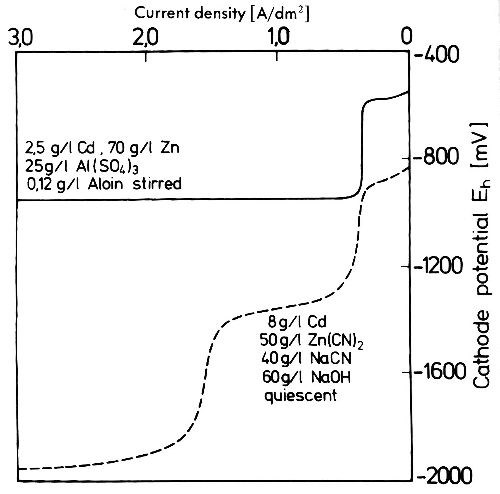
Figure 17 - Cathode potential-current density curves in quiescent acid and cyanide cadmium-zinc electrolytes at 20°C.
In cyanide solutions silver behaves as the most noble metal. Gold dipped into a cyanide silver plating bath dissolves without application of external current, and silver is deposited onto gold by electron exchange. Under normal conditions of practical silver electroplating, metals which form stable cyanide complexes are not discharged simultaneously with the silver. Gold and mercury are exceptions. In cyanide solutions gold is codeposited with silver at low current densities. If traces of sulfide ions are present, no codeposition of gold with silver will occur as long as the depolarization of the sulfide ion on silver is effective. As soon as the incorporation of the sulfide ion as silver sulfide in the silver deposit is complete and the cathodic potential has reached that of the sulfide free electrolyte, codeposition of gold begins as normal.
For the simultaneous deposition of other metals with silver from cyanide solutions it is of importance that the deposition potential of silver depends to a high degree on the molar ratio of silver to cyanide. At high cyanide concentration the predominant complex ion is Ag(CN)3-2; discharge of silver goes over to Ag(CN)2ˉ as the discharge determining step at a relative high potential. At lower cyanide concentration the normal Ag(CN)2ˉ exists, and the discharge of silver AgCN is decisive.9
Also under extreme conditions codeposition of less noble metals, which form stable cyanide complexes, such as iron, nickel and cobalt, is practically impossible.
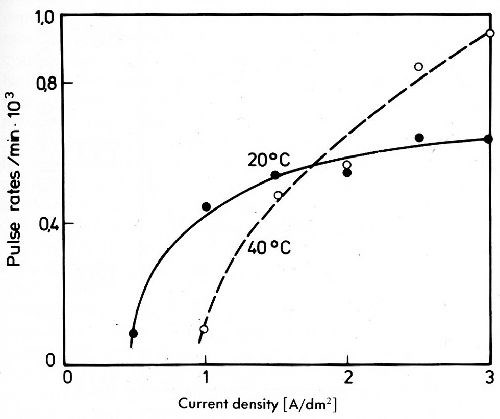
Figure 18 - Impulse rates of silver deposits from a cyanide electrolyte with 5 g/L Ag and 30 g/L Ni doped with 63Ni.
This is shown in Fig. 18 for an electrolyte containing 5 g/L silver, 30 g/L nickel as cyanide complexes and 80 g/L free potassium cyanide.39 Only at relatively high current densities can a very small amount of nickel be detected in the deposit by radioactive tracer method. It is not certain whether this nickel is really discharged or if it only results from electrolyte occluded by the rough surface of the silver deposited.
In electroplating practice nickel was used erroneously for a long time as a hardener for silver deposits. (It was proved that additions of nickel to the silver plating bath have no influence on the hardness of the deposits). But any codeposition of nickel must be excluded from consideration under normal conditions. Also, codeposition of copper and silver in a cyanide bath is possible only under extreme conditions, since the copper cyanide complex is much more stable than that of silver. In addition the influence of the cyanide concentration on copper is stronger than on silver. Therefore it is impossible to achieve codeposition of copper and silver at high cyanide concentration.
Zinc, cadmium and indium were repeatedly proposed as alloying metals for silver deposits to obtain tarnish resistant silver. Apart from the fact that these metals indeed prevent reaction of silver with sulfur, and thus tarnishing, they cause discoloring by the formation of oxide in a humid atmosphere. In cyanide alloy plating baths it is difficult to control deposition closely enough to get deposits with reproducible properties. It is possible to obtain deposits with zinc or cadmium content40 as shown in Fig. 19 for silver-zinc. The zinc content of the deposits rises very quickly when the limiting current density of silver is surpassed. At very low silver concentration of the electrolytes used this occurs at low current densities. As soon as the limiting current density of the zinc deposition is reached (between 1 and 2 A/dm2) a further increase of current density has no more significant influence on the composition of the alloy deposits. Only silver deposits with a low zinc content can be deposited in a usable form to higher thicknesses. Codeposition of silver and indium in cyanide solutions is possible too, if indium in the electrolyte is stabilized by dextrose which prevents indium hydroxide separation. In these baths indium shows the typical behavior of the less noble metal compared to silver. Besides problems in bath control silver-indium deposits tend to be discolored. They often show a typical spiral growth structure on the surface.41
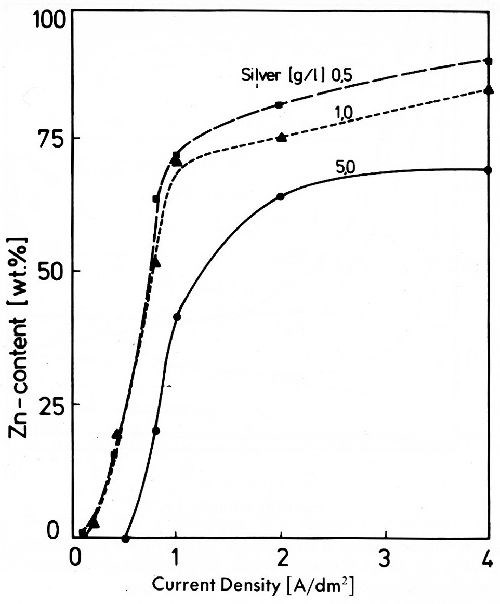
Figure 19 - Zinc content of silver-zinc deposits from on electrolyte with 100 g/L Zn(CN)2, 100 g/L NaOH, 160 g/L NaCN with silver contents of 0.5, 1.0 and 5.0 g/L.
The problem of tarnish resistant silver deposits by alloying metals is still unsolved.
Simultaneous deposition of other metals with silver can be achieved best with metals which form no stable complex cyanide ions, as lead, tin (as Sn+2 ion), antimony, bismuth or arsenic. To stabilize these metals in the alkaline cyanide solution, organic complexing agents, e.g., oxy-acids as citric or tartaric acid and other complex formers partly with alkaline hydroxide contents, are used.
Silver-lead alloys were proposed as friction bearings but in practice air craft engine bearings have successive layers of different metals deposited and then diffused by heat.
The advantage of silver bearings with lead-tin or lead-indium linings compared to cast copper-lead bearings is due to the fact that molten lead dissolves in silver during runs under extreme conditions. The microstructure of such a friction bearing shows lead segregations in the silver matrix. When copper-lead friction bearings are overheated the lead, not having alloyed with the copper, melts and collects at the overheated area, and blocks oil channels or holes.
Silver-antimony alloys with small amounts of antimony are generally used as hard and bright deposits. For electrotechnical purposes it must be kept in mind that the resistivity is considerably increased by the presence of antimony in solid solution with silver. By codeposition of arsenic it is easy to get bright silver deposits with a very good appearance but arsenic in these deposits is not quite insoluble in dilute acetic acid. A difficulty in the practical use of silver-tin baths is the tendency of Sn+2 in the electrolyte to oxidize to Sn+4. The latter cannot be codischarged.
In silver electrolytes which contain an alloying metal which does not form a stable cyanide, the composition of the alloy deposit can be influenced over a wide range by changing the cyanide content. Figure 20 shows curves of the bismuth content of deposits from silver-bismuth baths with equal silver and bismuth contents (25 g/L), and potassium cyanide concentrations between 22 and 150 g/L. At cyanide concentrations of 22 and 44 g/L, the bismuth content of the deposited alloys is very small. During electrolysis bismuth behaves as the less noble metal. With cyanide concentrations of 70 g/L and higher at low current densities silver-free bismuth is deposited. With rising current density the bismuth content of the deposits decreases quickly at first. After the limiting current density of the silver deposition is reached, it remains nearly constant. By changing the metal ratio in the electrolyte to higher silver contents the potassium cyanide concentration at which codeposition of bismuth is strongly favored, is changed to higher values.42
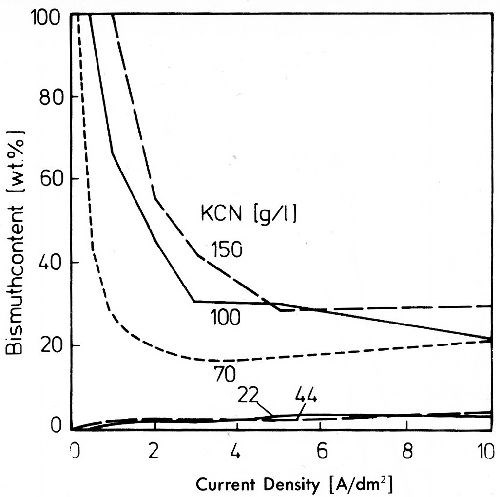
Figure 20 - Bismuth content of silver-bismuth deposits from a bath with 25 g/L Ag, 25 g/L Bi, 35 g/L potassium tartrate, 25 g/L potassium hydroxide and different cyanide concentrations, 25°C, stirred electrolyte.
Gold forms only one very stable cyanide complex of the composition Au(CN)2ˉ. In acid solutions the Au(CN4) complex of the Au+3 ion is relatively stable; in alkaline solutions it decomposes to Au(CN)-2, the complex of the Au+ ion, according to the equation.43
Au(CN)4ˉ ⇔ Au(CN)2ˉ + (CN)2
As far as is known, gold is the only metal to be discharged immediately via the Au(CN)2ˉ-complex predominant in the electrolyte. In comparison to other metals the influence of the cyanide concentration on the deposition potential is minor and the aforementioned stability in weak acid solutions is characteristic. From this many possibilities result, which influence the codeposition of other metals.
Gold alloy deposits of different compositions are used for jewelry and wares of daily use, as well as for technical applications, especially in the electronics industry. Not only binary but also ternary and even quaternary alloys are deposited.
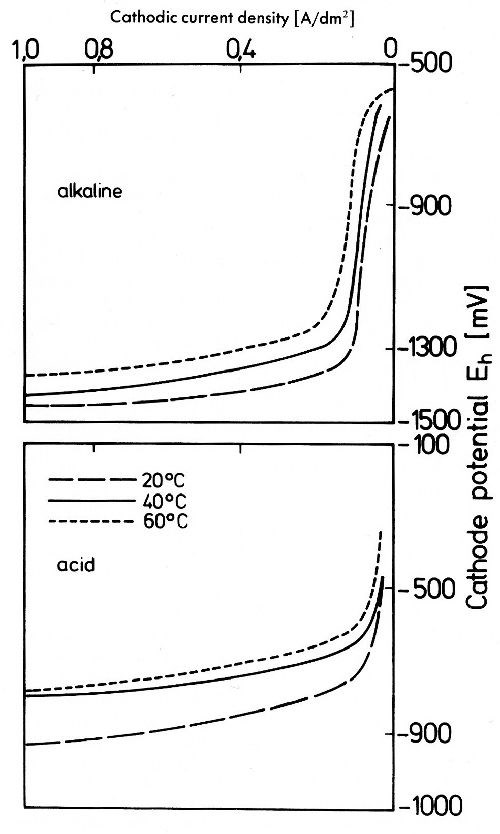
Figure 21 - Cathode potential-current density curves in gold electrolytes: Alkaline: 2 g/L Au as KAu(CN)2, 8 g/L KCN; Acid: 2 g/L Au as KAu(CN)2, 40 g/L citric acid, 60 g/L sodium citrate, pH = 4.2.
As Fig. 21 shows, the potential-current density curves for the alkaline cyanide baths are in general more negative than those in a weak acid bath, disregarding low current densities beneath the limiting current density range in the alkaline bath.44 Generally, current efficiency in acid electrolytes is much lower than in alkaline electrolytes, highly efficient acid solutions may exceed 55%. That in alkaline baths a limiting current density for the gold deposition exists is due to the mentioned fact that hydrogen is discharged in alkaline solutions immediately from the water molecule at a more negative potential; in acid solutions protons are discharged at a less negative potential.
The cathodic potential curves of a cyanide copper bath (Fig. 22) show a very strong dependence upon the cyanide concentration, and copper is finally no longer discharged. The codeposition of copper with gold therefore takes place much more readily in electrolytes in which the ratio of copper to cyanide is as high as possible. Figure 23 shows cathodic potential curves (upper part) and the metal contents of the deposits in relation to the current density (lower part) of a cyanide gold-copper electrolyte.

Figure 22 - Cathode potential-current density curves in potassium-cyanocuprate solutions with different ratios of CN:Cu; copper concentration 20 g/L, 60°C, quiescent electrolyte.
The potential curves show a marked limiting current density for gold deposition. As soon as this is exceeded codeposition of copper rises with the current density, at first very fast but soon levels out as a consequence of the exhaustion of the cathodic diffusion layer in dischargeable Cu(CN)2ˉ ions.
In acid baths, where only gold is present as a cyanide complex, codeposition of copper can be observed at very low current densities.
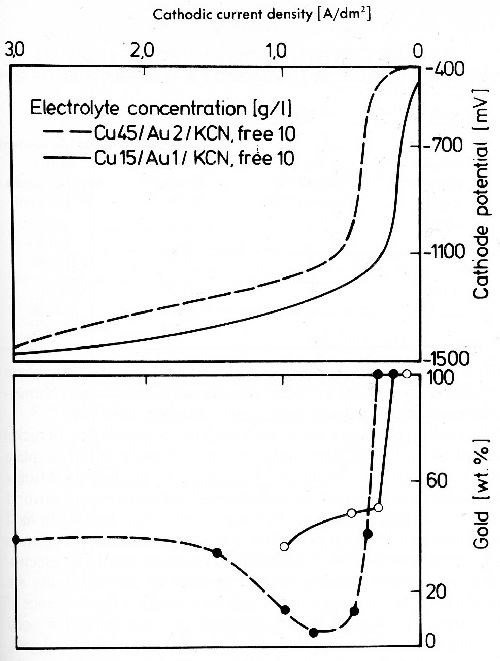
Figure 23 - Cathode potential-current density curves and gold content of gold-copper alloy deposits in stirred alkaline cyanide baths, 50°C.
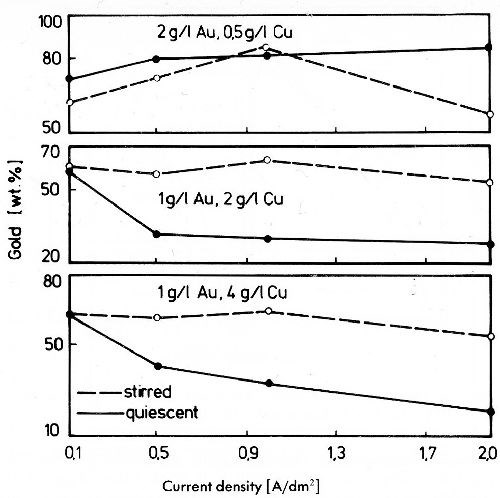
Figure 24 - Gold content of gold-copper alloy deposits from acid baths with different gold to copper ratio at 40°C and a pH of 5.5 (Au as KAu(CN)2, Cu as CuSO4, 60 g/L, sodium citrate, 40 g/L citric acid).
Figure 24 shows the changes of the gold content in deposits from acid solutions. In an electrolyte with low copper and higher gold concentration, the gold content of the alloys increases with increasing current density. At higher copper concentrations the gold content decreases with increasing current density. In these baths gold behaves as the more noble metal, and one might expect with rising polarization by higher current density the gold content of the deposits to get smaller. This contradiction can be explained by the fact that the electrolytes have relatively small concentrations of dischargeable mass particles. Therefore exhaustion will soon take place at the cathode, so that the discharge of gold or copper or both is diffusion controlled. In an electrolyte with only 0.5 g/L copper and 2 g/L gold, diffusion control for copper deposition starts at low current densities. At a copper concentration of 2 to 4 g/L and 1 g/L gold the diffusion control for gold begins at lower current densities. Therefore the gold content of the deposits decreases with rising current density.
Cobalt cannot be simultaneously discharged with gold in alkaline cyanide electrolytes.39 From an acid gold bath simultaneous discharge of cobalt with gold presents no difficulties (Fig. 25).45 At very low cobalt concentrations of the electrolyte codeposition of cobalt occurs. According to Fig. 25, cobalt content of the alloys decreases very quickly with rising current density due to the fact that cobalt concentration in the electrolyte is very small as compared to gold. This causes a diffusion controlled deposition.
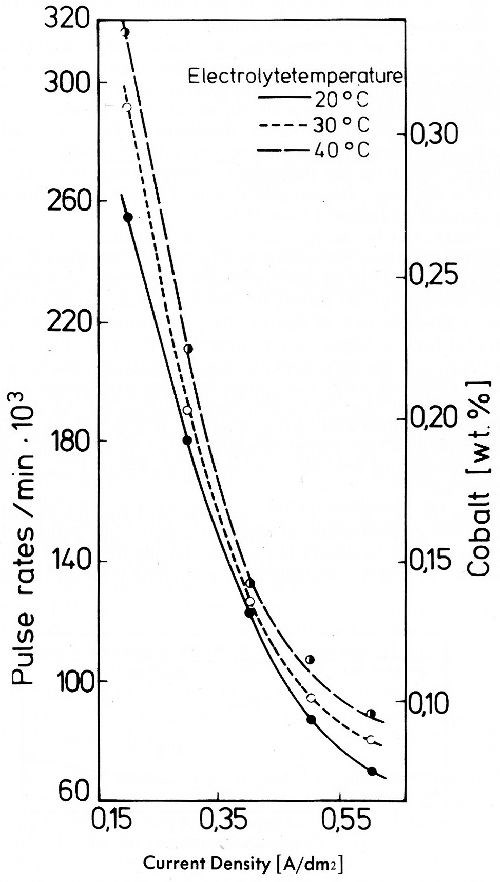
Figure 25 - Pulse rates and cobalt content of gold-cobalt deposits from a weak acid phosphate electrolyte with 120 g/L KH2PO4, 8 g/L Au as KAu(CN)2, 0.0136 g/L Co (doped with Co-60) as CoCl2, pH 4.5; 40°C.
Nickel may be codeposited with gold from alkaline and acid plating solutions. As shown in Fig. 26, the codeposition of nickel and gold in an alkaline cyanide bath starts as soon as the limiting current density of gold is reached. As seen from the right-hand part of Fig. 26, the codeposition of nickel is forced back to higher current densities45 by an addition of 1 g/L silver to the electrolyte. In an acid electrolyte which contains nickel, no longer as a cyanide complex, codeposition with gold starts at very low current densities (Fig. 27). As shown for an electrolyte with 2 g/L gold and 10 or 20 g/L nickel, alloy deposits with high nickel contents can be obtained. In acid electrolytes gold behaves as the more noble metal as compared to nickel.
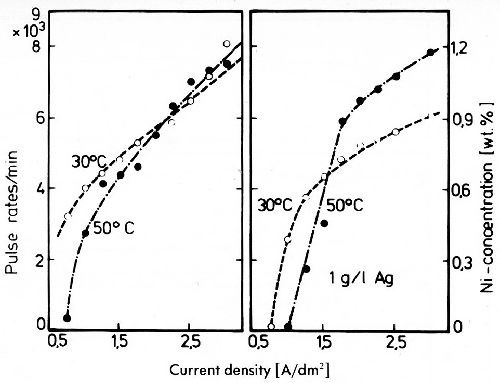
Figure 26 - Nickel content of gold-nickel deposits from an alkaline cyanide bath (10 g/L Au, 3 g/L Ni doped with 63Ni, 150 g/L KCN).

Figure 27 - Gold content of gold-nickel deposits from a citric acid bath with 2 g/L Au as KAu(CN)2, 10 or 20 g/L Ni as NiSO4, 60 g/L sodium citrate and 40 g/L citric acid; pH 4.2; 40°C.
For the production of the so-called hard gold alloys with only low concentrations of nickel or cobalt, alloy baths with relatively low concentration of the alloying metal are used. Cobalt- or nickel-containing hard gold alloys from acid baths sometimes show difficulties in soldering and contact problems in service caused by included non-metallic substances. Numerous investigations have been made into this problem.46-53
Inclusion is always associated with the codeposition of nickel or cobalt and gold. Even today it is not possible to fully explain the source of the inclusion. According to studies by Munier and others a polymer is codeposited. Quantity and ratio of carbon, nitrogen, oxygen, hydrogen and alkali metals in hard gold deposits make it probable that rather than a homogenous substance a mixture of different substances from the electrolyte is included. Among these, cyanides of low solubility are important. Interesting but not yet explained is the presence of hydrogen. Evidently it is contained as supersaturated solid solution, made possible by high lattice defects of the hard gold, or as occluded molecular hydrogen. Only part of it can be present in the form of chemical compounds. According to G.B. Munier,46 gold-silver deposits from an electrolyte with a pH of 11 show the same content of non-metallics as hard-gold deposits with nickel or cobalt from acid baths. This behavior can be explained by the fact that at low cyanide contents, the deposition of silver is connected with inclusion of AgCN as shown in Table 3.54 The danger of inclusion is not present when a sulfite bath is used, but this bath causes other difficulties, among them instability and often insufficient corrosion resistance of alloy deposits.20
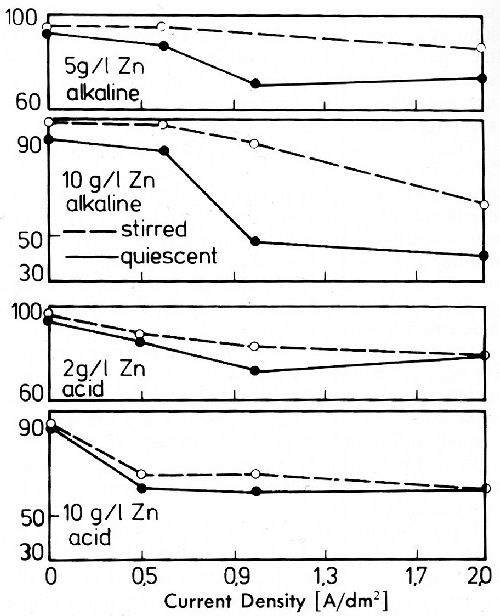
Figure 28 - Gold content of gold-zinc deposits: Alkaline bath: 2 g/L Au as KAu(CN)2, zinc as K2Zn(CN)4, 43 g/L KCN, 42 g/L KOH; Acid bath: 2 g/L Au as KAu(CN)2, Zn as ZnSO4, 60 g/L sodium citrate, 40 g/L citric acid, pH value 4.2; 40°C.
Figure 28 shows the gold content of alloy deposits as related to the current density for alkaline and acid gold-zinc alloy baths. Independent of the composition of the electrolyte and other working conditions, gold content decreases with rising current density.
The influence of the zinc content in the electrolyte, and of the transition from alkaline to acid bath on the composition of the alloys is slight. In alkaline and acid solutions gold is, at the investigated bath compositions, always the more noble metal. The zinc content in the deposits increases with increasing current density.44
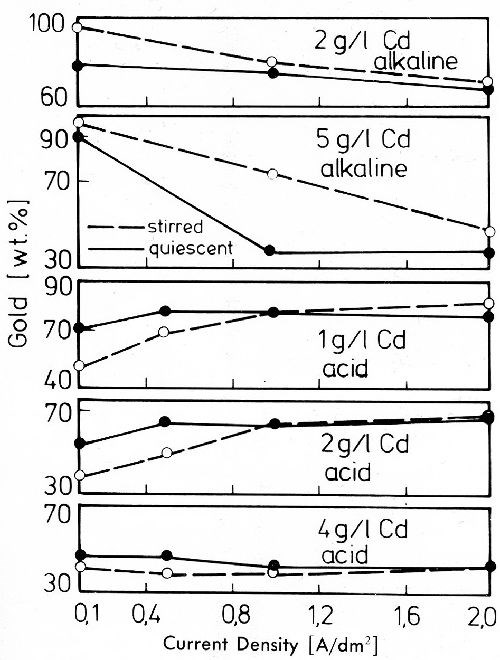
Figure 29 - Gold content of gold-cadmium deposits: Alkaline bath: 2 g/L Au as KAu(CN)2 and Cd on 5 g/L Cd as K2Cd(CN)4, 18 or 34 g/L KCN, 4 or 10 g/L KOH; Acid bath: 2 g/L Au as KAu(CN)2, Cd as CdSO4, 40 g/L citric acid and 60 g/L sodium citrate, pH = 4.2, 40°C.
Cadmium is of great interest as an alloying metal for gold; also for ternary and quaternary alloys. Contrary to zinc it is codeposited with gold at very low current densities below the limiting current density of gold alkaline cyanide electrolytes. As shown in Fig. 29, in the alkaline bath cadmium behaves as the less noble metal, the gold content in the deposits decreasing with rising current density. In an acid bath cadmium is more noble than gold. Its ratio to gold in the deposits is higher especially at low current densities.44 In alkaline as well as in acid baths it is possible to deposit gold-cadmium alloys in nearly every composition desired.
Metals such as lead and bismuth which form no stable cyanide complexes in alkaline solutions behave more nobly than gold. Their deposition is strongly preferred as shown for bismuth gold alloys in Fig. 30.55
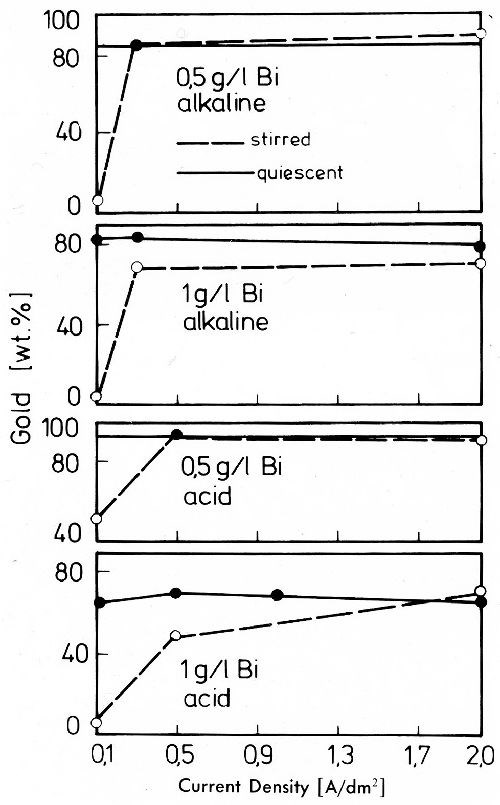
Figure 30 - Gold content of gold-bismuth alloy deposits from an alkaline cyanide both with potassium hydroxide and potassium tartrate to keep bismuth in solution and from a citric acid bath, gold content of both baths 2 g/L.
In acid cyanide baths besides cyanide which is present only in amounts enough to form the Au(CN)2ˉ complex a further complexing agent, such as citric acid, is added. Also ethylendiamintetraacetic acid (EDTA) is a stronger chelating agent for heavy metals, but the constant is different for various metals. It depends on the pH value of the solutions, as is also true of other complex compounds. The Au(CN)2ˉ complex itself is not influenced by EDTA significantly. The cathode potential curve is shifted to more noble values, but this is due to the depolarization of hydrogen deposition and not to that of gold.56 Nickel- and cobalt-EDTA complexes are very stable, and to discharge nickel it is necessary to keep the molar nickel to EDTA ratio above 1. For iron special conditions exist. The EDTA complex of the Fe+3 has a very high complex constant compared to that of Fe+2 so that in the presence of oxygen Fe+2 is quickly oxidized to Fe+3. Bright gold alloys with iron, nickel and cobalt may be deposited from acid baths in presence of EDTA as a third complexing agent in addition to cyanide and citrate. The optimum conditions for good deposits from such a solution are very limited.57 Of special importance is the molar ratio of metal to EDTA. The very narrow limits of the metal to EDTA ratio for crack free gold-iron alloys are shown in Fig. 31. The range of the optimum ratio of metal to EDTA changes with the metal codeposited with gold as given in Fig. 32.45
The current density range in which bright and crack-free deposits are obtained rises from nickel via cobalt to iron. For iron it is very high because it is necessary to exceed the limiting current density for the redox reaction Fe+3/Fe+2.
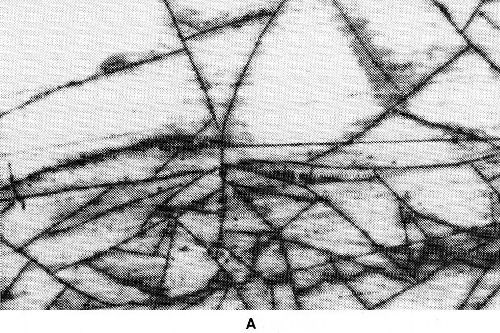
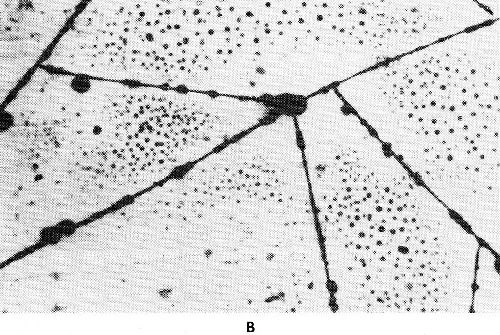
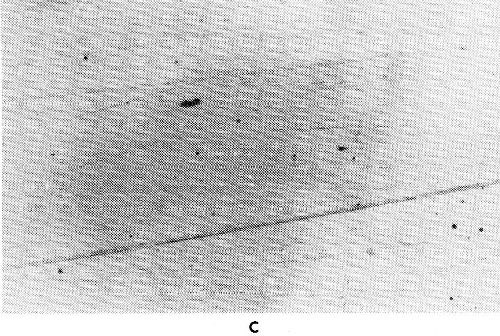
Figure 31 - Gold-iron alloy deposits from a citric acid bath with EDTA as third complexing agent; molar ratio Fe; EDTA (a) 1:0.95, (b) 1:0.8, (c) 1:0.7.
To possess sufficient chemical resistance gold alloy deposits should contain at least 75% gold. Often it is difficult to deposit alloys with controlled properties and at the same time keep their gold content within the desired tolerances. In alloys for decorative purposes with definite colors copper and cadmium are preferred to achieve yellow (Cd) or red (Cu) gold in different shades. Furthermore silver, zinc and in small amounts nickel are used. Nickel, cadmium and silver are generally contained in binary white gold deposits. For technical purposes binary gold-silver alloys are of interest. For special applications in the electronic industry other alloying metals, e.g., antimony, are employed.
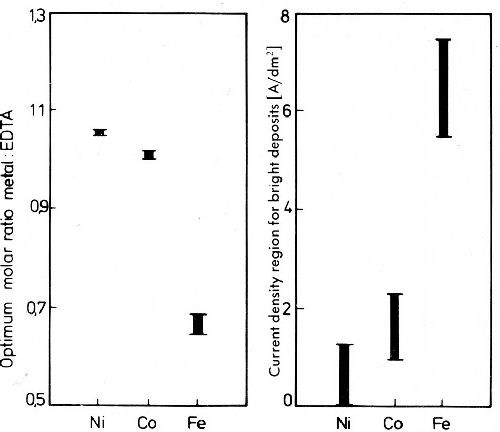
Figure 32 - Optimum conditions for the deposition of gold-alloy deposits with Fe, Co and Ni from the citric acid bath with addition of EDTA.
A comparison of properties of alloys, electrodeposited and melted and recrystallized
Electrodeposited alloys are usually not in a state of thermo-dynamical equilibrium.21,58 Electrocrystallization takes place in aqueous solutions at temperatures far below the temperatures at which detectable diffusion takes place. But if the deposition temperature is higher than that at which diffusion begins, stable structures are obtained. This occurs for alloys of low melting metals, such as tin-lead, tin-indium, zinc-cadmium and tin-zinc. Other alloys deviate in their properties more or less from cast alloys. This is due not only to the fact that they are not in equilibrium, but that their structure sensitive properties are often strongly influenced by inclusion of non-metallic substances. The structures of electrodeposited and cast alloys correspond inasmuch as, nearly always, only phases which exist in the phase diagram are formed. During electrocrystallization of alloys and crystallization of the casting, the components may crystallize separately, in the form of solid solutions or intermetallic phases. Great differences often exist in the concentration ranges of phases formed. Phases present in the phase diagram may be absent in electrocrystallized alloys, or phases which are unstable at the temperature of electrocrystallization may appear. Figure 33 gives, based upon x-ray studies, several more complicated phase diagrams of electrodeposited alloys in comparison to equilibrium diagrams. It can be seen that in part greater differences exist, but that on the other hand during electrocrystallization as well as luring solidification true alloys crystallize.
The differences in composition are of high importance to solid solutions. In spite of complete solid solubility between two metals, under certain circumstances very small or even no solid solution at all is observed in the electrodeposited alloys. On the other hand it is possible to electrodeposit alloys with an extended solid solution range when the phase diagram shows no remarkable mutual solubility of the bordering phases.

Figure 33 - Phase ranges of electrolytic and recrystallized alloys.
One must keep in mind that the structure of electrodeposited alloys may vary considerably with deposition conditions.
Microscopically, alloy deposits often show a characteristic structure. Solid solutions may have a laminar structure which can be caused either by periodic concentration changes of the soluted metal or by inclusion of non-metallic substances during electrocrystallization. Intermetallic compounds formed by electrocrystallization often show typical structures (Fig. 34).
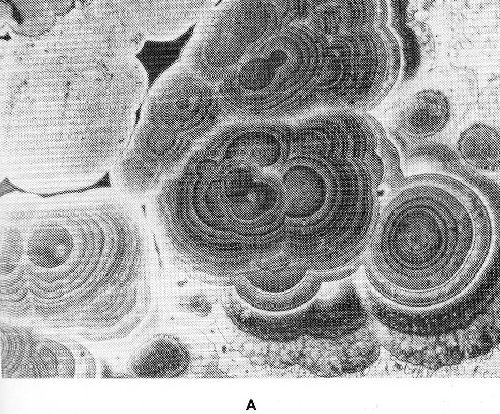

Figure 34 - Structure of brass deposits: (a) β-brass, 56.3% Cu, (b) η-brass, 11.7% Cu.
When a heterogenous mixture of two phases is formed during electrodeposition it may result in a very fine dispersion. The phases can also crystallize in a very coarse form as shown by Fig. 35. Coarsely crystallized deposits are of no practical interest unless the intercrystalline cohesion is strong.

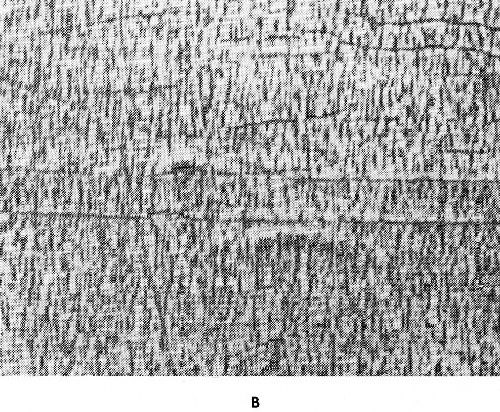
Figure 35 - Structure of cadmium-silver deposits: (a) 37% Cd (deposited from a simple cyanide electrolyte), (b) 40% Cd (deposited from a silver containing bright-cadmium electrolyte).
The different behavior of two metals during simultaneous electrocrystallization may be explained on the basis of the cathodic potential current density curves.
Dependent on the electrolyte composition and the metals present, codeposition of the less noble metal may occur only after exceeding the limiting current density of the more noble metal. In this case either a mixture of both metals or of the more noble metal and an intermetallic compound is formed. The intermetallic compound must approach the electrochemical properties of the less noble metal. However, if no limiting current range separates the simultaneous discharge of both metals then crystallization by formation of solid solutions may occur during electrodeposition, even when no solid solution in the phase diagram exists. In this way, highly supersaturated solid solutions form, which normally cannot be produced in cast alloys or only by very fast quenching. One disadvantage is that electrolytic crystallization of these highly supersaturated solid solutions is nearly always associated with inclusions of non-metallics.
An influence by inclusion upon the extreme supersaturation achieved during electrodeposition of alloys cannot be denied. It is known that soluted atoms concentrate at sites with high lattice defects. By inclusion, extremely high lattice distortions are produced which facilitate supersaturation.
Hydrogen as a alloying element
In discussing the structure of electrodeposited alloys hydrogen is often forgotten despite the fact that it is the most common alloying component, stated by Blum and Hogaboom 25 years ago. On cadmium and zinc surfaces the overvoltage of hydrogen is very high. In the baths of these metals codeposition of hydrogen occurs after the limiting current density is exceeded.38 Electrodeposited cadmium and zinc do not contain hydrogen in supersaturated solution. All hydrogen discharged escapes as molecular hydrogen. The hydrogen in electrodeposited zinc is formed by a reaction between zinc and water or zinc hydroxide, which is occluded in the deposits, according to:59
Zn + H2O ⇔ ZnO + H2 and
Zn + Zn(OH)2 ⇔ 2ZnO + H2
These reactions proceed very slowly at room temperature. As hydrogen does not diffuse in zinc, it is enriched during longer storage at room temperature, as shown in Fig. 36.
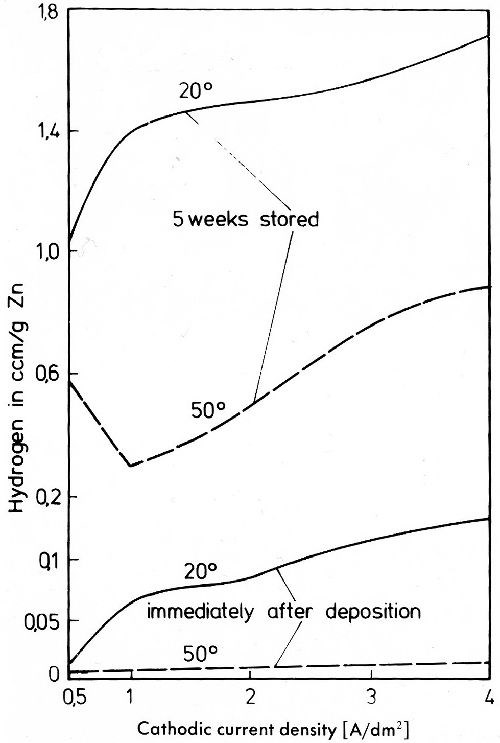
Figure 36 - Hydrogen content of zinc deposits determined by cold extraction.
To investigate this behavior of zinc deposits it is necessary to use the method of cold extraction of hydrogen by amalgamation. During hot extraction the reaction of zinc with water or zinc hydroxide is very fast, so that at a sufficiently high extraction temperature it will be completed in a short time. The total hydrogen evolved by these reactions is easily determined by high temperature extraction. Figure 37 gives the ZnO content after extracting, the hydrogen evolved and the molar ratio of zinc oxide to hydrogen plotted against current density for zinc deposits from cyanide baths. The slopes of the curves for zinc oxide and hydrogen evolution are equal. The pronounced minimum in the curves is caused by the limiting current density of zinc. The molar ratio of zinc oxide to hydrogen increases continuously from a value of nearly 1.0 at low current densities to about 1.75 at a current density of 4 A/dm2. If the total amount of hydrogen were formed with water according to the first equation the molar ratio would be 1:1 and in the case of the second reaction with Zn(OH)2 it would be 2:1. In no case is the ratio less than 1, which would make possible the conclusion that at least a certain part of hydrogen evolved during crystallization is trapped in voids as molecular hydrogen. The proportion to which each of the two possible reactions of zinc and water or zinc hydroxide participates in the formation of hydrogen changes with current density. At increasing current densities the inclusion of zinc hydroxide increases. Zinc cyanide is not included by zinc from cyanide solutions. This confirms that the deposition does not take place via a cyanide complex as the discharge determining step but via zinc hydroxide.
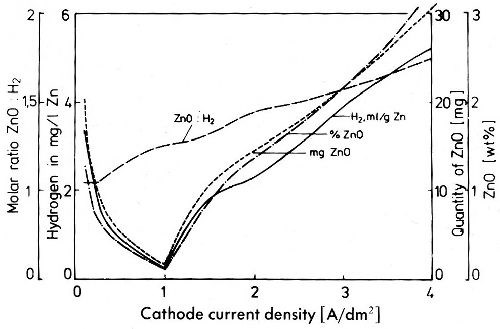
Figure 37 - Hydrogen and zinc oxide contents of zinc deposits from a cyanide electrolyte as determined by hot extraction.
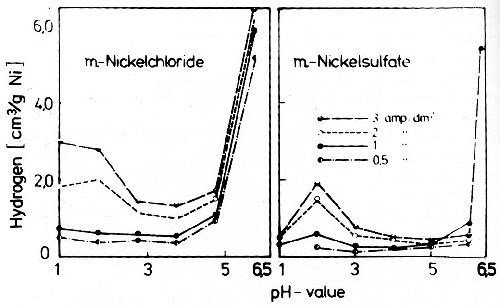
Figure 38 - Relationship of the hydrogen content of nickel deposits to the pH value of the electrolyte. Electrodeposition temperature 20°C.
Very high supersaturation of dissolved hydrogen is observed in metals which are not deposited without simultaneous hydrogen discharge, such as iron, cobalt, nickel, chromium and platinum metals. Figure 38 shows the influence of pH value on the supersaturation of nickel with hydrogen deposited in chloride and sulfate solutions.60 The hydrogen content is low at a low pH, where the current efficiency for the discharge of nickel is lowest, and where the hydrogen discharge is the highest. But at high pH-values where the simultaneous hydrogen discharge is lowest, hydrogen supersaturation is very high, as seen from Fig. 38. At very high values, over a pH of 5.0 in the chloride bath and over a pH of 6.3 in the sulfate bath, the hydrogen content of the nickel jumps. The reason for the extreme increase in the hydrogen content is the hydrolysis of nickel salts at these pH values. The nickel chloride hydrolysis constant is higher than that of nickel sulfate, therefore hydrolysis followed by adsorption and inclusion of basic nickel chloride occurs at a lower pH. The high lattice defects caused by inclusion of the hydrolysis products lead to very strong enrichment of hydrogen. Strains in the nickel lattice causing high hydrogen supersaturation may also be formed by codeposition of a second metal, which enters into a solid solution with nickel. But it is not certain that adsorption and inclusion of other non-metallic substances do not co-operate, which is absolutely possible, e.g., in nickel-zinc or nickel-thallium alloys. Figure 39 shows the influence of the zinc concentration in electrodeposited zinc-nickel solid solutions on their hydrogen content. The hydrogen supersaturation limit of the face-centered cubic nickel-zinc solid solutions increases very fast till the saturation limit of the zinc is reached. At higher zinc concentrations, hydrogen content of alloys again decreases and reaches zero at about 80 wt% zinc.

Figure 39 - Hydrogen content of electrolytic face centered cubic zinc-nickel solid solution in relation to the zinc content of the alloys.
Detailed x-ray structure analysis showed that in chloride solutions hexagonal nickel by hydrogen codeposition61 is not formed.62 In contrast a chromium hydride of the composition CrH with hexagonal structure has been confirmed.63 The formation of this hydride during the electrocrystallization of chromium depends on the electrolysis conditions. It can be deposited in pure state with pulsating DC at temperatures below 25°C. The formation of chromium hydride is favored by high current densities and high chromic acid concentrations. The hexagonal hydride possesses only a small homogeneity range from CrH0.95 to CrH1.00. The existence of a hydride with face-centered cubic structure is confirmed, yet it has not been possible to establish its composition. It is believed that the face centered cubic hydride is formed from the hexagonal hydride by an alteration of the stacking order of the more densely packed lattice planes. In chromium hydrogen alloys the body-centered cubic chromium lattice along with the hexagonal lattice is seen in a wide concentration range from Cr-H0.04 to CrH0.95.
Supersaturated hydrogen-nickel solid solutions decompose in air at room temperature slowly with the evolution of hydrogen. CrH too, is an unstable compound, but in air it does not decompose at room temperature. This stability occurs in the same way as the passivity of chromium, by the formation of a pore-free atmospheric oxide layer which prevents hydrogen evolution. Chromium hydride decomposes in vacuum at room temperature slowly; in a few minutes in air slow deposition begins at 100°C. The chromium remaining after low temperature decomposition of the hydride is very active. In air it forms a thicker oxide layer with accompanying heats, yet oxidation soon stops.63 This type of chromium has a more noble potential than electrolytic chromium.64 The properties of chromium hydride are similar to those of normal electrolytic chromium.
Properties of electrodeposited solid solution
As has been discussed, included non-metallic substances are of high importance to the formation of highly supersaturated nickel-hydrogen alloys. The same behavior was observed in the formation of some highly supersaturated binary solutions of two metallic components. As examples, alloys of silver with lead, bismuth or thallium and of copper with lead may be mentioned. In these systems very highly supersaturated silver or copper rich alloys are deposited from electrolytes of appropriate composition. Probably complexes with organic oxy-acids; e.g., tartrate, citrate or, in the case of copper-lead alloys, a copper(I)-thiourea complex, are included. That true solid solutions are formed may be seen from the lattice constants of the solvent metal. The lattice constant is a property of low sensitivity to lattice strains and it is possible to hold the inclusion of foreign substances during electrocrystallization low enough, so that the x-ray interferences do not fade into the background too much. As long as no films or filaments of the included substances exist, only a very small increase in the resistivity by lattice defects will be observed. Normally in solid solutions it increases very fast. As shown in Fig. 40, the resistivity of electrolytic copper-lead alloys rises very quickly in the range of copper rich alloys up to about 12 wt% Pb. Since copper does not dissolve in lead, the resistivity changes continuously from pure lead to the supersaturated copper rich solid solution. According to the phase diagram the maximum of the solid solution of lead in copper, 0.1 wt%, is reached at 900°C and 0.04 wt% at 500°C.65
The solid solubility may be different for the same metals, depending on the composition of the electrolyte. This has been stated for silver-cadmium40 and copper-gold44,65 alloys. Depending upon the bath composition, these alloys crystallize in a wide range as solid solutions or in two phases. In the alkaline cyanide electrolyte, simultaneous discharge of copper and gold begins above the limiting current density of gold. In nearly neutral, or more so in weakly acid solutions in which codeposition of copper with gold occurs at lowest current densities, copper crystallizes in the formation of solid solutions with gold.44,66
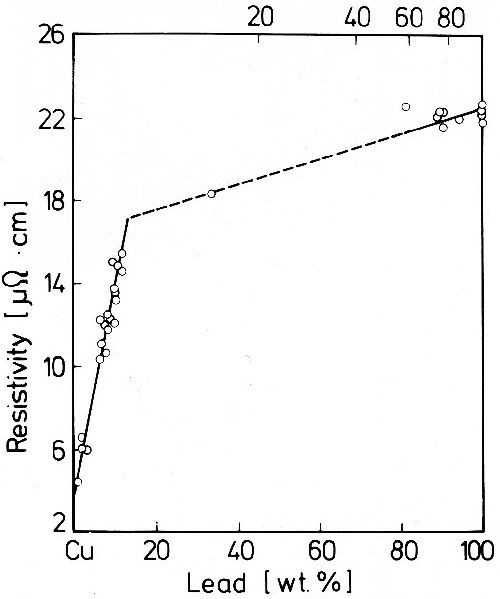
Figure 40 - Resistivity of lead-copper deposits.
The supersaturated electrodeposited alloys show strong deviations from supersaturated solid solutions of cast and recrystallized alloys after quenching.
Supersaturated quenched silver alloys of lead or bismuth have only a hardness of about 40 kp/mm2. Electrodeposited alloys of silver-lead or silver-bismuth have hardnesses of 140 to 190 kp/mm2, depending on the plating conditions. Copper-lead alloys may reach a hardness of 300 kp/mm2. It is striking that there is no significant hardness relationship with the concentration of the soluted metal. Furthermore the hardness equals that of metals with inclusion of non-metallics without the codeposition of a second metal.
The electrodeposited alloys can be brought into thermodynamic equilibrium by heat treatment. Changes of their properties during annealing are different from those of recrystallized supersaturated alloys.
Characteristic changes in resistivity are given in Fig. 41. Electrodeposited supersaturated solid solutions show no age hardening. Segregation temperature and rate are independent of the atomic supersaturation. The fact that, as shown in Fig. 41, for silver-lead and silver-bismuth alloys resistivity passes through a minimum, and increases at higher temperatures, is due to the increase in solubility with increase in temperature.
Hardness, caused by lattice defects independent of dissolved metals, shows another behavior. It does not change during the segregation process. This is shown in Fig. 42 for a silver-bismuth alloy. The same behavior was observed much earlier for solid solutions of hydrogen in nickel and chromium.67 Hydrogen can be evolved without any change in hardness of the metal under consideration.
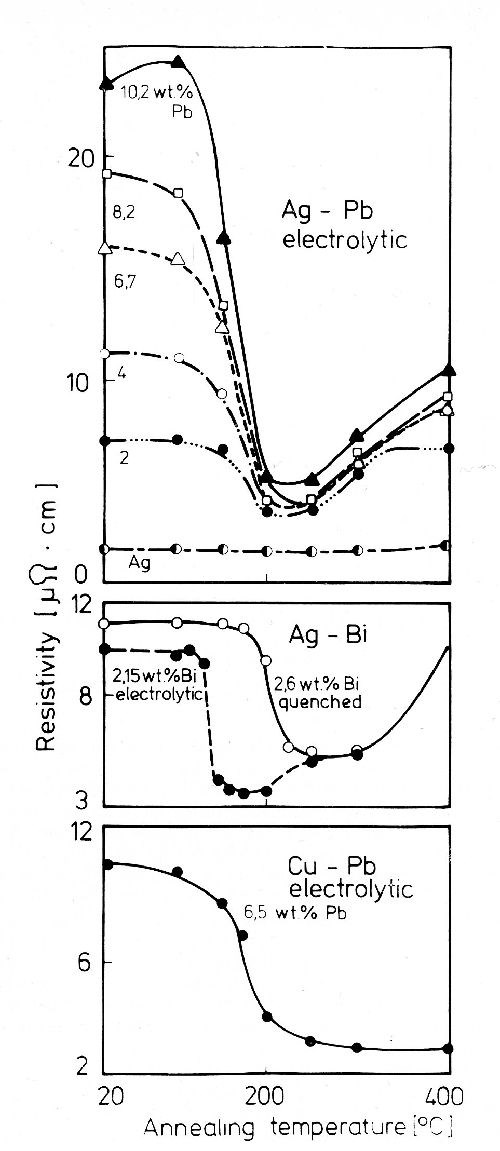
Figure 41 - Resistivity changes of supersaturated alloy deposits during aging.

Figure 42 - Changes of the properties of a bismuth-silver alloy deposit with 2.15 wt% Bi during aging at 140°C.
Heat treatment of electrodeposited alloys
By heating it is possible to achieve equilibrium conditions in all electrodeposited alloys. Included inorganic or primarily organic compounds are decomposed during heating, and develop gases which lead to porosity. At higher inclusion levels, e.g., electrolytic lead-copper alloys can decompose to powder) during heating. At lower inclusion levels, they get porous and brittle.
Hard gold alloys from acid gold baths contain inclusions as already discussed. After heating at higher temperatures such deposits show a surface with blisters and craters as shown in Fig. 43. Generally it can be stated that alloys with inclusions are not suitable for technical applications at elevated temperature. Thus it is not possible to get from copper-rich lead-copper alloys with a hardness of about 300 kp/mm2, an alloy with an as-cast hardness of about 40 kp/mm2 through heat treatment. Electrodeposited lead-copper alloys treated in this way are brittle and not suitd for friction bearings. In contrast copper-poor lead alloys are well suited for linings of aluminum friction bearings, since no solid solution and no troublesome inclusion occurs.
Electrodeposited alloys free from detectable amounts of inclusion show no formation of pores or blisters during heating. When their structure corresponds to that of the phase diagram, all properties behave normally during annealing. After a critical temperature is reached a sudden decrease in hardness and tensile strength connected with a discontinuous increase in elongation occurs, as shown for an 18-carat gold-copper-cadmium alloy by F.I. Nobel, D.W. Thomson and J.M. Feibel.68 Such electrodeposited alloys offer the same problems as cast alloys of the same composition for use at higher temperatures.
A heat treatment is often of special value for practical use when during simultaneous electrocrystallization little or no solid solutions are formed. Electrodeposited gold alloys are only chemically resistant when all components, at a high enough gold content, are present in solid solution with gold. This is significant for white and colored gold alloys, containing metals like copper, cadmium or nickel, when these metals are not or are only partially in solid solution and crystallize as less noble metals or gold compounds. If in these alloys the second phase is present as small particles, it is easy to bring them into solid solution with gold at adequate temperatures. The necessary temperature depends on the specific metals to produce solid solution with gold. The heat treatment should be in vacuum or in a neutral or reducing atmosphere to prevent oxidation. At most in colored gold alloy deposits from alkaline cyanide baths the inclusion of non metallics is so slight that no difficulties will occur during heat treatment.

Figure 43 - Crater formation on the surface of a hard gold deposit (Ni-Au) from the acid bath after heating at 500°C for 15 min. Original magnification 10,000×.

Figure 44 - Diffusion in copper-plated zinc after annealing at 400°C for 24 hr.
Diffusion properties are of interest to the electroplating industry from other points of view too. Diffusion in the solid state takes place only when the components form solid solutions or intermetallic compounds. Low-melting metals in which diffusion occurs at room temperature or lower are able to form alloys at rather low temperatures. The so called "sink in" of brass deposits on zinc is caused by diffusion of zinc to the surface, which assumes the color of zinc. Also between silver or copper deposits and underlying zinc, diffusion is fast at relatively low temperatures. This property was utilized for the production of brass deposits with controlled composition by electrodepositing copper and zinc successively on iron with a subsequent heat treatment. Using this method one has to keep in mind that first copper must be deposited on iron, because during heating, diffusion and alloy formation between zinc and iron occurs as well as between copper and zinc. If zinc is first deposited it is impossible to get a brass layer with desired properties. When one of the components diffuses faster than the other, holes may form in the diffusion zone (Kirkendal effect). This is shown in Fig. 44 for copper deposits on zinc after annealing at 400°C.
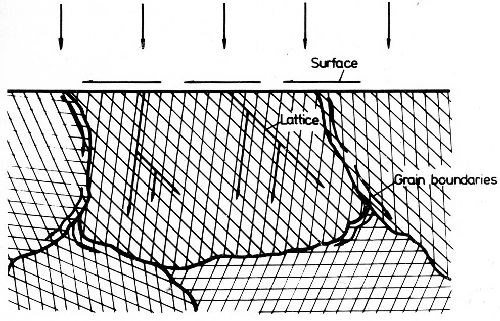
Figure 45 - Schematic representation of the diffusion types.
The diffusion between surface layer and underlying metal is of great interest. Under certain circumstances it can be of practical advantage, in other cases it is disadvantageous. One has to distinguish between surface, lattice and grain boundary diffusion, as shown in Fig. 45. Surface diffusion is characterized by atoms diffusing along the surface. During lattice diffusion they move uniformly along within the crystal lattice. Grain boundary diffusion along the grain boundaries is generally undesirable. Figure 46 shows grain boundary diffusion for a pure gold deposit on silver. By this type of diffusion the effective layer becomes thinner. The effect of diffusion of gold on alpaca, a nickel-copper-zinc alloy, is quite different. Figure 47 shows in the left portion gold plated alpaca prior to diffusion annealing. In the right portion the same sample is shown after diffusion treatment. In this case a thicker alloy plate has formed by lattice diffusion, which is still corrosion resistant. Only small grain boundary diffusion occurred. A sharp distinction between the three types cannot be made. In general, all three types of diffusion can be observed simultaneously, e.g., in chromium-iron samples.
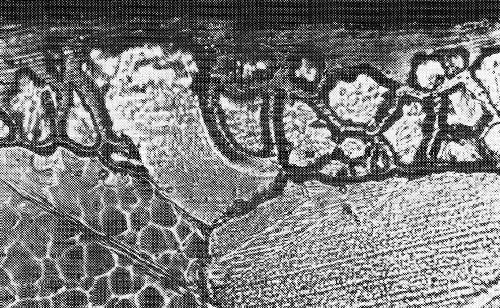
Figure 46 - Diffusion of gold and silver in gold-plated silver during annealing.

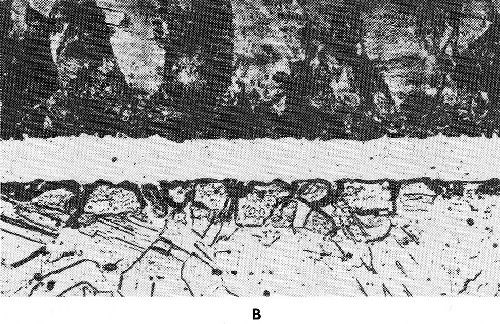
Figure 47 - Gold-plated alpaca with 13% Ni: (a) prior to diffusion annealing, (b) after diffusion annealing.


Figure 48 - Surface diffusion in sheet steel, chromium plated on one side: (a) annealed at 850°C, 24 hr., (b) annealed at 1000°C, 24 hr.
As an example of surface diffusion, the diffusion of chromium and iron, may be mentioned. Figure 48 shows the edge of an iron sheet chromium plated on one side after annealing at 850°C and 1000°C. On the original chromium-free edge a thin chromium-iron alloy has been formed at 850°C. Annealing at 1000°C has caused a relatively thick chromium-iron alloy layer on the unplated surface. Surface diffusion of chromium on iron depends on the atmosphere. In the presence of only small amounts of oxygen, surface diffusion is nearly zero. Trace amounts of volatile chlorides, such as chromium (II) or iron (II) chloride in the atmosphere strongly intensify surface diffusion. An intermediate layer of copper retards the diffusion of the chromium and iron as long as the temperature is below the melting point of copper. Most influential is the carbon content of iron because of the formation of ternary chromium-rich carbides. These act as a barrier layer and prevent further diffusion.
The pronounced surface diffusion of chromium on iron is sustained by the relatively high vapor pressure of chromium at high temperatures.
To prevent diffusion, nickel is used under gold layers on silver or copper and copper alloys. But for very thin layers of gold, nickel as barrier layer is not sufficient since at temperatures high enough the gold-nickel diffusion has to be considered. According to J.C. Turn and E.L. Owen,69 chromium and nickel with 8 to 10% P are the most effective barriers for the copper-electrodeposited gold system.
High melting point metals, such as molybdenum, tungsten, tantalum and others react readily with gases, especially oxygen, at high temperatures. This behavior restricts their usage. To prevent these reactions electrolytic surface layers of precious metals have been proposed. As protective coatings, silver deposits cannot be used because oxygen diffuses in silver at elevated temperatures at a very fast rate, so that oxidation of the underlying metal occurs even if the silver layer is free from pores. Gold and platinum are the only metals which are impermeable for gases at high temperatures. Gold does not alloy with molybdenum or tungsten. With other high melting metals, such as vanadium, niobium and tantalum alloy formation takes place at relatively low temperatures and the function as barrier layer is lost. Gold can be used in an extended temperature range only on molybdenum and tungsten, but a disadvantage is that pores in the gold deposit on these metals cannot be sealed by heat treatment. Furthermore, at temperatures near the melting point of gold very coarse grains are formed and at the grain boundaries of the gold layer pores show up through which oxygen reacts with the underlying metal. Platinum reacts with all high melting metals in the solid state, and intermetallic phases are formed.
Since gold does not diffuse into molybdenum or tungsten, it was of interest to investigate the influence of an intermediate layer of gold before electroplating with platinum. Platinum and gold diffuse at high temperatures very rapidly with the formation of mutual solid solutions. The affinity of platinum to the high melting metals to be protected is higher than it is to gold. Platinum diffuses through gold and reacts with molybdenum forming the hexagonal close packed ε-phase of the platinum-molybdenum phase diagram.70 At high enough temperatures, the cubic A-15 phase is formed which exists only as a high temperature phase. Its presence causes embrittlement and crack formation in the alloy layers. After annealing long enough, all of the platinum migrates through the gold, so that finally a pure gold layer is on the surface and beneath it molybdenum-platinum phases are formed. The structures of these phases depend on annealing temperature and time. Therefore platinum and gold-platinum double layers, as protection for high temperature use of high melting metals, are restricted by temperature and time.
As shown, the formation of alloys by diffusion of a deposited metal with the underlying metal, or by mutual diffusion of two or more electrolytic layers may be useful in some cases but in others it is detrimental.
Summary
In the present paper only some aspects of alloy plating could be discussed. In a broader sense the problems of the electrodeposition of alloys are very complex and touch nearly all questions of electrodeposition. In the past years alloy plating has grown fast, especially for functional purposes. It must be understood that alloy plating, sometimes in connection with diffusion treatment, is only used when it gives advantages over pure metals. In spite of enormous research and development work, not all possibilities of alloy deposition for practical use have been exhausted. The properties of electrodeposited alloys are quite often different from those of melted alloys of the same composition. This is sometimes a disadvantage and prevents immediate industrial use but shows new directions for further scientific and practical work.
Not all alloys in technical use can be electrodeposited. On the other hand electrolytic deposits of alloys which have no importance in the cast and recrystallized state are used technically, e.g., nickel-tin.
Abner Brenner, in his book Electrodeposition of Alloys, on page 256 called me "a protagonist of the idea that the properties of even the bimetallic alloys are largely determined by the presence of inclusions." You will understand that living up to this reputation in my paper, non-metallic inclusions were discussed extensively.
Acknowledgements
The investigations on electroplating discussed in this lecture, as far as they concern work done in the Forschungsinstitut fur Edelmetalle und Metallchemie Schwäbisch, Gmund, have been sponsored over long years by the Deutsche Forschungsge-meinschaft, the Ministry of Economic Baden-Württemberg, the Arbeitsgemeinschaft Industrieller Forschungsvereinigungen, the Deutsche Forschungsgesellschaft für Blechverarbeitung und Oberflächenbehandlung and other agencies. I want to take this opportunity, to thank all of them.
Last not least: I thank my friend Mr. R. Scott Modjeska, who corrected the manuscript meticulously in order to enable the readers to appreciate what I wanted to say.















Related Content

How to Maximize Nickel Plating Performance
The advantages of boric acid-free nickel plating include allowing manufacturers who utilize nickel plating to keep up the ever-changing regulatory policies and support sustainability efforts.
Read More
Advantages to Pumped Eductor Agitation
Not all agitation methods are created equally. Pumped agitation with eductor nozzles can improve process tanks and quickly show a reduction in operating costs while keeping staff safe, following environmental legislation and preventing pollution.
Read More
3 Tests to Ensure Parts are Clean Prior to Plating
Making sure that all of the pre-processing fluids are removed prior to plating is not as simple as it seems. Rich Held of Haviland Products outlines three tests that can help verify that your parts are clean.
Read More
How to Choose Between Sulfate and Chloride-Based Trivalent Chromium
There are several factors to consider when choosing between sulfate and chloride-based baths for trivalent chromium plating. Mark Schario of Columbia Chemical discusses the differences and what platers should keep in mind when evaluating options.
Read MoreRead Next

Episode 45: An Interview with Chandler Mancuso, MacDermid Envio Solutions
Chandler Mancuso, technical director with MacDermid Envio discusses updating your wastewater treatment system and implementing materials recycling solutions to increase efficiencies, control costs and reduce environmental impact.
Read More
Delivering Increased Benefits to Greenhouse Films
Baystar's Borstar technology is helping customers deliver better, more reliable production methods to greenhouse agriculture.
Read More
Education Bringing Cleaning to Machining
Debuting new speakers and cleaning technology content during this half-day workshop co-located with IMTS 2024.
Read More