
Electroplating, Electrochemistry and Electronics - The 15th William Blum Lecture - Part 3
This article is the third of four parts of a re-publication of the 15th William Blum Lecture, presented at the 61st AES Annual Convention in Chicago, Illinois, on June 17, 1974. Dr. George Dubpernell reviews the history and extent of commercial plating, then delves into the electrochemical science, including potentials, overvoltage and connections to electronics.



by
George Dubpernell



M&T Chemicals
Ferndale, Michigan
Recipient of the 1973 William Blum
AES Scientific Achievement Award
Editor’s Note: Originally published as Plating & Surface Finishing, 62 (6), 573-580 (1975), this article is the third of four parts of a re-publication of the 15th William Blum Lecture, presented at the 61st AES Annual Convention in Chicago, Illinois, on June 17, 1974. A printable PDF version of Part 3 is available by clicking HERE. The printable PDF version of the complete 44-page paper is available HERE.
Hydrogen overvoltage in electroplating
The hydrogen overvoltage of the basis metal being plated is often a dominant factor in plating from both acid and alkaline solutions, and may even determine whether or not any plate at all is obtained. For example, cast iron, malleable iron, or high carbon steel surfaces may become difficult or impossible to plate after cleaning and pickling in preparation for plating, due to the low hydrogen overvoltage of the resultant surface. Such surfaces of pickled or acid dipped cast iron or high carbon steel rival the surface of platinized platinum used for the hydrogen electrode in having a low hydrogen overvoltage, making it so easy to evolve hydrogen at a low potential that it is difficult to deposit many metals.
Alkaline cyanide zinc solutions often could not be used for plating cast iron or malleable iron parts, as only hydrogen evolution was obtained, or at best the coverage was poor.104,105 One solution to the problem was to introduce a little mercury into the bath, or to dip the part in an acid mercury-containing dip prior to plating in order to increase the overvoltage of the surface, and this procedure was patented.106-108 It was also found that treating such surfaces in boiling hot, concentrated cyanide solution for 5 to 30 min would increase their overvoltage and permit them to be zinc plated.109 Sometimes the only remedy is to grind or polish off the low-overvoltage surface, or to plate over it with a heavy undercoating of another metal such as copper or cadmium. Two more recent references show that this type of behavior is still a problem.110,111
Chromium plating utilizes very strongly acid baths, and is only possible on bright, dense, highly-polished surfaces in general (which have a relatively high hydrogen overvoltage). This was clearly recognized by Haring and Barrows in 1927.112 If nickel surfaces are "passive," it may be impossible to chromium plate them. This can happen in a number of ways. Anodic alkaline cleaning tends to oxidize nickel and make it "passive," and should be avoided. Poorly-covered zinc die castings may have dull, streaky, dark nickel deposits in the recesses which are difficult to cover with chromium and even tend to prevent coverage on surrounding areas. The use of black nickel plate to produce low overvoltage electrodes has been patented.113
Bright nickel plate from solutions containing excessive amounts of brightener may be "passive" and impossible to chromium plate even though it appears satisfactory. Probably very small invisible amounts of nickel compounds on the surface are sufficient to give a low overvoltage condition, under the reducing conditions at the cathode. Similarly, Raney nickel powder is a catalyst used for hydrogenation, and very small quantities are effective.
The theory of the electrodeposition of chromium itself is probably very closely tied up with the theory of hydrogen overvoltage. A black, powdery coating of chromium probably has a low overvoltage and tends to inhibit further chromium deposition and promote hydrogen evolution. The success of the hexavalent chromium plating solution may be due to the ability of chromate to complex or otherwise render harmless such impurities as iron, copper, zinc, nickel, lead, etc.
This type of impurity is difficult to control in trivalent chromium baths. The removal of lead is a definite step in the operation of chromium ammonium sulfate baths used for the electrowinning of the metal,114,115 and, if such baths are shut down for more than a few minutes, a low overvoltage condition results on the cathode chromium surface which may take many hours of electrolysis to overcome. This period of electrolysis is probably in the nature of an electrolytic purification, and is also required in breaking in a new bath before the current efficiency can be brought up to the normal level of around 50% or more.
The current efficiency in chromium deposition is closely related to the hydrogen overvoltage of the surface and simultaneous hydrogen evolution, although 10 or 20% or more may also be lost in side reactions such as reduction of hexavalent to trivalent chromium, or of trivalent to bivalent. If the temperature of the solution is increased, the current efficiency is generally affected quite strongly.
Figure 23 from a report by N.E. Ryan116 shows typical behavior of 300 g/L chromic acid solutions catalyzed with sulfate and with fluoride additions, and electrolyzed at over 1000 A/ft2 for chromium production. It seems likely that the low current efficiency for heavy deposits in boiling hot solutions with sulfate is due to a low overvoltage condition, whereas in fluoride-catalyzed solutions a cleaner, more oxide-free surface is maintained which probably has a higher overvoltage and gives a higher current efficiency. Shluger and Mikhailova117 have shown that the oxide film on sulfate-catalyzed chromium deposits contains even more sulfate proportionately than is present in the solution, while Shluger and Osbenkova118 felt that there was no oxide film at all on fluoride-catalyzed chromium deposits.
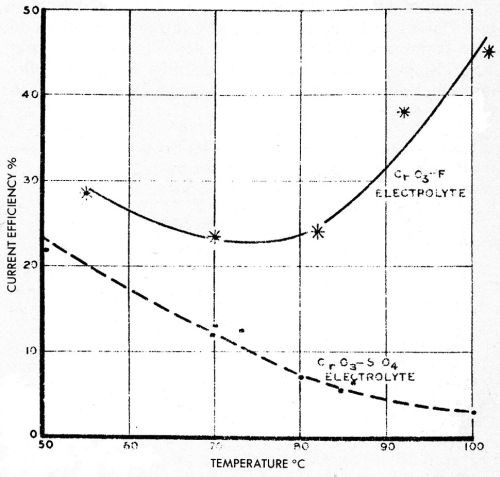
Figure 23 - Ryan’s curves of current efficiency of chromium deposition at high temperatures with sulfate catalyst and with fluoride catalyst. Reprinted by permission from the Commonwealth of Australia, Aeronautical Research Laboratories, Melbourne.
Low overvoltage is not necessarily a problem or a hardship in all cases in chromium plating. Thus, Bedi119 used immersion deposits of platinum or palladium on steel to secure a low overvoltage condition for etch prevention at low current density areas in fluoride-catalyzed baths, and Bedi and Dubpernell120 used the same surface treatment or even etched cast iron121 to prevent chromium deposition at high current densities for stop-off purposes. Glover122 reported an investigation of chromium plating on cast iron in which he found that high phosphorus seemed more important than the carbon in making cast iron difficult to plate.
In trivalent solutions, Jangg and Burger123 obtained current efficiencies as high as 68% on a mercury cathode in chromium ammonium sulfate solution at pH 3.2. However, in many tests they obtained low current efficiencies due to the formation of black, spongy deposits with low overvoltage. Some of their results for chromium sulfate and chromium chloride are given in Figs. 24 and 25, and show the rather unusual result of decreasing current efficiency with increasing concentration. This should presumably be explained as due to impurities that cause lower overvoltage deposits in more concentrated solutions, but have less effect in more dilute baths.
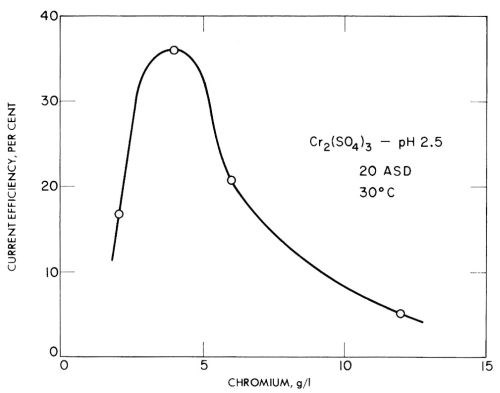
Figure 24 - Jangg and Burger's measurements of the current efficiency of chromium deposition on a mercury cathode from chromium sulfate solution. Plotted with permission from Pergamon Press.
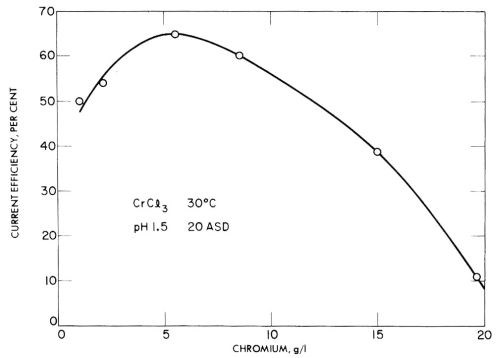
Figure 25 - Jangg and Burger's measurements of the current efficiency of chromium deposition on a mercury cathode from chromium chloride solution. Reprinted with permission from Pergamon Press.
J.E. Bride and co-workers have obtained a number of patents on chromium plating from trivalent solutions which give some indication of the dominant effect of impurities and overvoltage in these solutions, particularly if the plating speed results reported are recalculated as current efficiencies. Some of these results from one of the patents124 are given in Fig. 26, and show the lack of reproducibility of three supposedly identical solutions tested at slightly different temperatures and made up at different times. This is presumably due to varying impurity effects and resultant varying overvoltage, as is also the falling off of current efficiency with current density. Another way of saying it might be to say that probably injurious impurities are complexed to a varying extent according to the details of the procedure and the exact organic additive used.
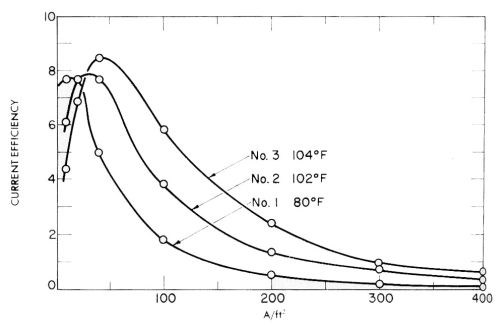
Figure 26 - Huba and Bride's measurements of the current efficiency of chromium deposition in three newly made up identical trivalent chromium plating baths at different temperatures.
Contact potential - Volta potential - Electrostatic surface potential
A continuous controversy has been going on under the name "contact potential" from the time of Volta right down to the present day, something like 180 years. Patai and Pomerantz125 and Langmuir126 state that Volta first discovered and measured the effect in 1797. When Volta disclosed the voltaic pile in 1800 (Ref. 1, p. 42), the title of his paper was "On the Electricity Excited by the Mere Contact of Conducting Substances of Different Kinds." Volta hoped that he had discovered a means of perpetual motion, but time has not borne him out. It appears from another source (Ref. 127, pp. 133-134) that both Volta and Pfaff had worked out the first tables of the elements in the order of their ability to excite electricity, in 1793.
The name "contact potential" seems something of a misnomer, since the metals are first placed in contact but then separated for measurement of the contact potential or Volta potential across a thin, dry gas space, or in a vacuum. For this reason, the name "electrostatic surface potential" has been included above as being more descriptive. It was employed by W.A. Zisman, and seems worthy of future use (Ref. 128, pp. 1-9, "The Solid/Liquid Interface - An Essential and Active Frontier of Science").
Some of the confusion connected with contact potentials is related to their magnitude. This is often thought of as being in the range of μV, or at most a few mV, and thus unimportant in most work. That this is not true is easily evident from Fig. 27, showing the change in mercury surface potential in dry nitrogen upon addition of 2 ppm zinc (Ref. 92, pp. 219-226, paper by A.H. Ellison, G.A. Lyerly and E.W. Otto on "The Effect of Impurities upon the Surface Potential and the Spreading of Liquids on Mercury"). Within about a minute, the zinc appears at the surface of the mercury and changes the surface potential with reference to gold by about 0.80V. When the surface of the mercury is swept, it wrinkles up, and purification by removal of a surface film appears to result, as the surface potential changes considerably in the direction toward clean mercury.
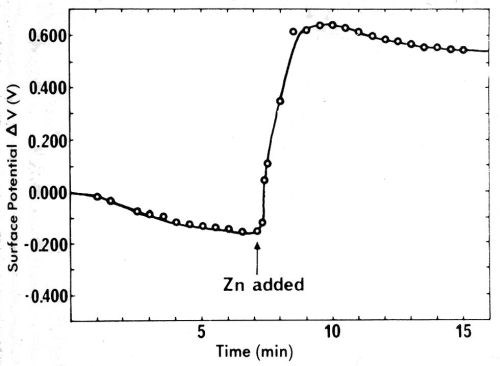
Figure 27 - Ellison, Lyerly and Otto's measurements of the change in mercury surface potential upon addition of two ppm zinc. Reprinted with permission from Marcel Dekker, Inc.
Contact potentials are fairly reproducible in a number of dry gases and in a vacuum, and the exact distance between the electrodes is not critical. Generally, they are spaced 0.5 mm (0.0156 in.) apart at first, and moved further apart during the measurements. There is a great similarity to measuring a single electrode potential against a reference electrode, with the electrolyte in between replaced by a dry gas or a vacuum.
Lord Kelvin in 1897-1898129 reviewed the previous controversies, pointed out that measurements of contact potentials as high as almost 4 V had been made, and described a dependable method of measurement. Figure 28 taken from Zisman130 shows the type of apparatus employed. If A is moved away from B, it causes a deflection of the quadrant electrometer. If a potential is applied to counterbalance the contact potential, a point is reached where moving A away from B no longer gives a deflection of the electrometer, and thus gives a determination of the contact potential of A measured against B as a reference electrode.

Figure 28 - Zisman’s diagram of the most commonly used method of measuring contact potential differences. Reprinted by permission from the Review of Scientific Instruments.
A good recent review of contact potential measurements is given by A. Dravnieks (Ref. 131, pp. 236-239). Dravnieks discusses the difficulties of these measurements and the choice of reference electrodes. He gives an interesting table of values based upon Michaelson’s132 tabulation of work function measurements. This is reproduced in Table 1 with the work functions in a vacuum from which it was derived, and with the values for sodium and potassium from the same source added. The contact potentials are calculated using gold as the best available reference electrode, and by subtracting the work function of gold from that of each of the other metals. The familiar electromotive series is given for comparison. It is difficult to avoid the conclusion that these are merely two different methods of measuring the same quantity. Using gold as the reference electrode for the contact potentials tilts the noble metal end of the scale by about a volt or more, but the close relationship is still obvious.
Table 1
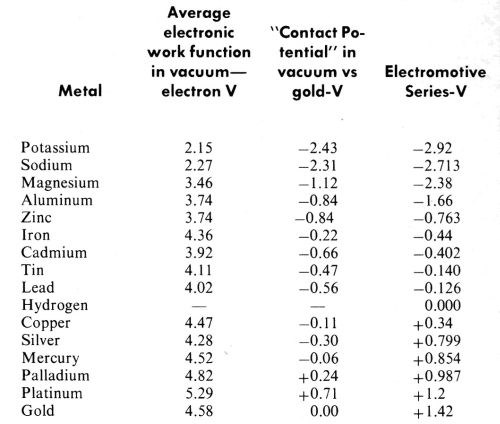
While the contact potential measurements are not ordinarily of great accuracy and reproducibility, the potentials of the electromotive series have a somewhat similar background and are largely calculated from the thermodynamic properties of materials rather than being measured values (Ref. 51, p. 88; Ref. 68). Contact potentials or electrostatic surface potentials are quite sensitive to adsorbed layers on clean metallic surfaces, and this property has led to their use in studies of adsorption, such as those by Zisman and his collaborators128 for example.
Instead of using a vacuum or a dry gas, Fort and Wells133 found that steady, easily reproduced contact potential difference measurements could be made in a saturated nonpolar organic liquid environment such as cyclohexane or benzene. Using electrodes submerged in such media they were able to study the effect of the distance of separation of the electrode from the gold reference electrode. They found that contact potentials could be measured to a precision of ±0.001 V in organic liquids.
An even greater accuracy of ±0.000001 V useful in semiconductor work was found by Steinrisser and Hetrick,134 using a low voltage electron beam technique to determine the photovoltage or photoelectric emission of cadmium sulfide exposed to an intermittent beam of light in a high vacuum.
Langmuir126 was convinced that contact potentials and the electromotive series were intimately related, and stated "The Volta series of the elements is determined by the magnitude of ϕ, the electron affinity" (electronic work function). On the basis of the limited data available to him in 1916, he concluded that "... contact electromotive forces can account for only about half of the observed electromotive forces of cells." One might say that the more recent measurements given in Table 1 indicate an even better correlation.
A number of subsequent workers have studied this inter-relationship, but invariably appear unable to conclude that the two types of potentials are an identical property of the elements arrived at by different methods of measurement or calculation. Gurney135 in 1932 showed the parallelism between the electrochemical series and the Volta contact potential series on the basis of general theoretical and mathematical considerations, and concluded that interface potentials are due to the electrons at the metal-metal junction, and that the Nernst theory of solution pressure of the metals is incorrect. He continues these same views in a later book (Ref. 136, pp. 33-38, 192-200).
Klein and Lange137-139 made their own measurements of Volta potentials of the elements against mercury as a reference electrode, in an argon atmosphere. However, they do not seem to compare these measurements with the electromotive series, and instead devote their attention to contact potential measurements between mercury and aqueous solutions. Even in a book in 1962,140 a chapter is devoted to the Volta potential without discussing any possible relationship to the electromotive series.
Butler141 makes a comparison in 1951 on the basis of zinc as the zero reference electrode, and concludes that the metal contact potential difference is a large part of the electromotive force of the cell. He does not quite seem to consider these forces identical, but "... the electromotive forces generally show a fairly close parallelism with the contact potentials."
On the basis of the theory developed in the present paper, it appears that "contact potentials" or electrostatic surface potentials are largely eliminated when pieces of metal are pressed together in good electrical contact in the external circuit. All of the pieces of metal in the exterior circuit are combined as if consisting of a single piece of metal; the conduction electrons of each piece of metal are free to circulate among all of the pieces of metal and establish their own equilibrium. Now only the exterior surfaces of each piece of metal are free to exhibit the intrinsic "contact potential" of that metal, and it would seem best to regard this "contact potential" as having been largely eliminated at or near the points where the pieces of metal are pressed together. At or near these latter points where the pieces of metal are pressed together, only smaller differences of potential remain such as the contact resistance (IR drop), thermoelectromotive force, etc.
The high electrical conductivity of metals apparently does not mean that all points in a given piece of metal are at the same potential. It is a common observation that a piece of metal which is being chromium-plated may only plate on the projecting parts, and the low current density areas may receive no chromium plate at all. This means that during the flow of current, at least, the projecting areas were at a higher cathode potential, and the recesses at a lower cathode potential, simultaneously.
It is not good practice to plate articles made up of a number of pieces of different metals stamped or welded together. If this has to be done, the first effort is to find a solution, perhaps cyanide copper, which will cover all of the metals in the assemblage with a good coating of a single metal, such as copper. Thereafter, the piece can be plated easily as a single piece of metal. Failing this, one expects difficulty around seams and welds between the different metals, or part of the assembly may plate readily and other parts not at all.
The so-called hydrogen electrode
It appears that any metal coated with its oxide exhibits an electrode potential behavior sensitive to acid and alkali that can be used to measure acidity and alkalinity over certain ranges of pH. Such responses have already been indicated in Figs. 5, 6, and 7 for tin, chromium and platinum. Others are shown in Fig. 4. Antimony and palladium are sometimes used. The glass electrode can possibly be considered a silicon oxide electrode. Mercury-mercury oxide electrodes are used for accurate pH measurement in alkaline solutions.
Bates (Ref. 142, pp. 304-306) describes a number of other "hydrogen ion electrodes," including tungsten, tellurium, bismuth, iridium, rhenium, osmium, ruthenium, and manganese dioxide. Ives and Janz (Ref. 143, pp. 116-117) also discuss hydrogen electrodes based on other metals, and include gold, rhodium, nickel and selenium, in addition to many of the above. New hydrogen electrodes are patented from time to time. One recent patent described uranium oxide,144 and another palladium oxide.145
A number of electrodes are quite far removed from the hydrogen electrode and yet give good responses to acidity and alkalinity. The mercury-mercury oxide electrode and the silver-silver oxide electrode are 0.926 and 1.17 V, respectively, above the hydrogen electrode, and are useful in alkaline solutions (Ref. 143, pp. 334-335). Cahoon146 found the manganese dioxide electrode to be about 1.07 V above the hydrogen electrode, and to give an accurate pH response from pH zero to pH 11.5 with the apparent possibility that it could be used beyond this range. Cahoon has also contributed a good practical article on the use of reference electrodes.147
While a hydrogen atmosphere is generally employed in the operation of the platinized platinum hydrogen electrodes and a few similar electrodes, most of the other metal-metal oxide reference electrodes mentioned above are used in air and have no special requirement.
Two notable investigations containing a great deal of information on the above matters were published in 1928 by French and Kahlenberg148 and Closs and Kahlenberg.149 In the first paper, French and Kahlenberg148 report the changes in electrode potentials of 24 metals and graphite, starting with air-saturated normal potassium chloride solution in each case, when oxygen, nitrogen or hydrogen was bubbled through the solution. Nitrogen and hydrogen generally had similar effects, and air and oxygen were often similar. Other gases studied in some cases were carbon monoxide, methane and helium. The effect of stirring and complete submersion was also examined. In general, the gases had very pronounced effects on the potentials of the metals.
In the second paper, Closs and Kahlenberg149 showed that a number of metals could be used instead of the hydrogen electrode for the potentiometric titration of acids and bases, particularly molybdenum, tungsten, arsenic, antimony and bismuth, but also tin and aluminum. While the metals have different potentials at the start, they all give the same end-point in an acid-base titration followed potentiometrically. In the discussion to this paper Professor Kahlenberg said: "I think that the other electrodes here mentioned are just as reproducible as the hydrogen electrode. They can be calibrated and used, any of them, particularly those that are not attacked. I do not believe we need a hydrogen electrode at all; ..."
The writer would conclude from the widely varying potentials in the above work that the so-called hydrogen electrode is only one example of a number of electrodes displaying similar behavior, and that its potential does not seem to have been shown to be that of hydrogen at all.
Dr. Blum characterized Kahlenberg's work in one of the above discussions very well when he said (Ref. 148, p. 194) "Dr. Kahlenberg has a faculty - I do not know whether I would call it a happy faculty or not - of developing a lot of facts which cannot be explained. I am not going to try to explain them."
The Nernst theory of the electromotive activity of the ions
Arrhenius proposed the theory of electrolytic dissociation in 1883-1887, and Nernst the theory of the electromotive activity of the ions in 1889.150 Both of these theories seem to have been rather highly overworked, and it would appear to be time for serious reconsideration and re-evaluation. The objections would seem to have most commonly been ignored, brushed aside, and lost sight of.
Glasstone (Ref. 151, pp. 18-20, 321-322) gives a brief review of some of the objections to the theory of electrolytic dissociation. There was also a well-timed and fairly comprehensive symposium on the subject in September, 1903, at the fourth meeting of the American Electrochemical Society in Niagara Falls, New York. The situation was well summarized by Bancroft,151a and an extensive discussion followed.
One of the strongest opponents was Professor Louis Kahlenberg (1870-1941) of the University of Wisconsin. Strangely enough, Kahlenberg studied in Ostwald's laboratory in Leipzig where he received his Ph.D. in 1895. He was at first an enthusiastic supporter of these new theories until he found that they did not agree with many experimental facts, and vigorously opposed them from about 1900 on.152
A comprehensive bibliography of the publications of Kahlenberg and his associates is available.153 In a few years, Kahlenberg's position became clear and was stated in four articles in 1901-1905,154-157 in addition to many research reports. One remarkable feature of his research reports was that many of them were published in the Journal of Physical Chemistry, even though its editor, W.D. Bancroft (1867-1953) was devoted to the theory of electrolytic dissociation. Lincoln152 quotes Bancroft as writing to Kahlenberg: "You are right, Kahlenberg, but you will find that you cannot convince people with facts."
Apparently this proved to be true. On the occasion of the 25th anniversary of the theory of electrolytic dissociation, G.N. Lewis158 discussed many of the facts showing what a poor basis the theory had, and yet concluded in favor of the theory on the basis of its "serviceability."
Kahlenberg practiced what he preached. He wrote a text book of general chemistry159 which made no use of the ionic theory and replaced it instead with the concept that solutions were merely chemical compounds having variable proportions. His textbook was popular and was used for quite a period of time, and passed through several editions. Kahlenberg established an enviable reputation as an excellent teacher.
However, Kahlenberg's opposition to the electrolytic dissociation theory was largely to no avail at the time. Lewis45 promoted the activity coefficient which made some allowance for the behavior of individual ions, but was not adequate for much improvement, as has already been discussed (sometimes called a waste-basket factor, in that it included many errors and variables). The Debye-Huckel interionic attraction theory was introduced in the 1920s to extend the range of concentration covered by the theory of electrolytic dissociation, but did not extend to high concentrations or make an extensive alteration of the circumstances. Kahlenberg said despairingly in the discussion to LaMer's paper in 1927: "The trouble is, we don't take our Epsom salts seriously enough!" Unfortunately, this expressive comment was toned down in the printed discussion.160
The same volume of the Electrochemical Society Transactions included a paper by Bancroft161 which reviewed some of the difficulties with the Nernst and Arrhenius theories. Outstanding is his comment that the measured pH of a solution does not necessarily show anything about the actual concentration of the hydrogen ion. Bancroft gives an example of the addition of enough sodium chloride to 0.1N HCl making the apparent hydrogen ion concentration equivalent to 0.5N HCl, and that this would correspond to a dissociation of 300% or more, which is, of course, absurd. This type of effect is still under study.162,163
One might say at this point, what about the burgeoning field of ion selective electrodes, as described by Frant,164 for example? In general, these electrodes do not serve to determine a given ion directly in the plating bath. Commonly, the desired ion is diluted very extensively in a medium of constant background composition and pH, and the desired ion may itself be altered in the process. Great skill and remarkable manipulative variations have been used to seek out "Nernstian" conditions and responses in this area of development, but it is obvious that electrode potentials are more often than not non-Nernstian. European workers165 have expressed a preference for the term "ion sensitive" rather than "ion specific" or "ion selective."
The Pourbaix potential-pH diagrams166 provide a useful means of studying and recording thermodynamic data. If one considers that ions in solution are not electromotively active or potential-determining, but, at most, merely modify the pre-existing electrode potential, which is an intrinsic property of the electrode material, then it is of interest to examine the significance of the most general type of potential-pH diagram. Figure 29 is such a diagram (Ref. 167, p. 112; also Ref. 166, p. 100, and Ref. 168, p. 36). The line "a" is the potential of the hydrogen electrode, which also is the basis of the pH scale (Figs. 2 and 3), and the line "b" is the oxygen electrode. Potentials above the line "b" correspond to anode reactions and potentials below the line "a" correspond to cathode reactions. These reactions can be produced chemically as well as electrolytically, as by using hexavalent chromium, permanganate, hydrogen peroxide, fluorine, or other oxidizing agents to get oxidation or an anodic effect; and bivalent chromium, hydrosulfite, hypophosphite, metallic sodium or other reducing agents to get reduction or a cathodic effect.
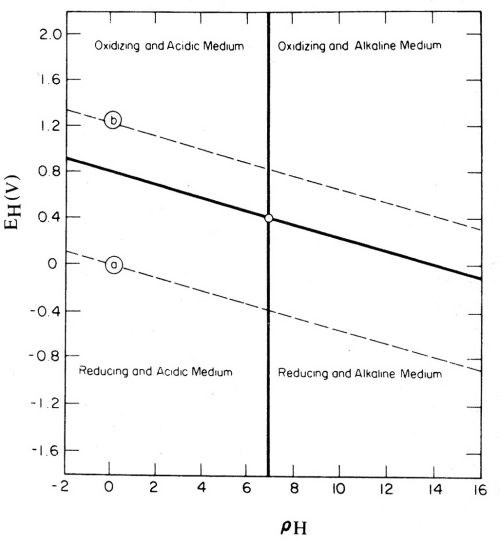
Figure 29 - Pourbaix diagram of electrochemical equilibria in water containing acid, alkaline, oxidizing and reducing media. Reprinted with permission from Plenum Publishing Corp.
If the potentials in the diagram are not a measure of ion concentrations, it becomes a question as to what determines them. Are we measuring the concentration of the two different carriers of electricity at the interfaces, the electrons and positive holes? It would seem that acid and alkali involve the absence or presence of excess electrons, and oxidizing and reducing the presence or absence of positive holes, or vice versa. Jorgensen48 considers that acid-base equilibria have to do with proton activities, and redox equilibria with electron activities. One wonders if acidity and alkalinity are merely a special form of oxidation and reduction, or are there four different carriers of the electric current?
Electrophysiology
Electrochemistry appears at least partly as an outgrowth of electrophysiological studies, and electrochemists have always been fascinated by the obvious importance of electrical phenomena inherent in living organisms. According to Motte-lay (Ref. 169, pp. 298-300) studies of electric fishes and eels were made in the 16th, 17th and 18th centuries prior to Galvani's work on "animal electricity" which was first published in 1791.1,170 John Hunter, a distinguished Scottish surgeon, received the Copley Medal of the Royal Society in 1775 for his work on the dissection of electric fish.171 Hunter found as many as 1182 "columns" or separate layers ("electroplaques" or "electroplates" in French) in each of the two electric organs of a very large fish 1.3 m (4.5 ft) long and weighing 33 kg (73 lb.) (Ref. 169, pp. 240-241).
It was the background of Galvani's work, and also that of Ritter,2 that stimulated Volta to discover the voltaic pile or battery. Volta did not like ascribing an animal origin to electricity, and showed that it could be generated by metals in contact with salt solutions.
The electric eels of the Amazon River are said to be capable of delivering shocks of up to 600 V and one A or more, sustained for several milliseconds, in the course of killing or stunning their prey172-174 (also, Ref. 175, p. 157). The source and handling of this much electric power in nature does not seem to have received adequate explanation.
Beutner176,177 proposed a theory based upon the contact potential differences of the two sides of the biological membrane which appears to have merit. Grundfest (Ref. 175, pp. 141-166) considers the source of bioelectric potentials to lie within the membrane itself.
Dr. G. W. Crile (1864-1943) was a pioneer surgeon who devoted much attention to the electrical nature of life. With his associates, he was said to have dissected some 3734 animals of all sizes and varieties.178 He published many books and articles, but the titles of two seem particularly suggestive.179,180
A great deal has been published in this general area, and a comprehensive review is not possible in the present article. Several additional titles seem particularly noteworthy.181,182
The Electrochemical Society, Inc. has continued activity in this field with the publication of a recent symposium.183 The first article in this symposium is a noteworthy review of electrochemical events in tissue growth and repair by A.A. Pilla (Ref. 183, pp. 1-16). Pilla calls attention to the remarkable work of R.O. Becker on the stimulation of bone growth by extremely low current density cathodic treatment similar to plating except for the very low current density.184 Figure 30 shows the result of Dr. Becker's tests over a seven day period.
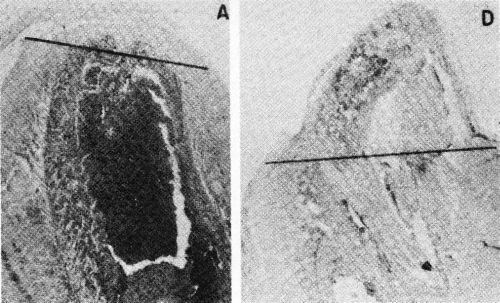
Figure 30 - Becker's results on the stimulation of bone growth of amputated rat's legs after seven days. The line shows the approximate level of original amputation. A is the control, D shows growth due to stimulation of extremely low current density as cathode. Reprinted with permission from Macmillan Journals Limited.
Relationships to electronics
Many relationships to electronics have already been made evident. In 1796, (Ref. 185, p. 4) Volta named all dry conductors, conductors of the first class, and all wet conductors or liquid conductors, conductors of the second class. He had apparently originated this terminology a few years earlier (Ref. 127, pp. 132-133) in connection with the description of the table of metals and other conductors of the first class, in the order of their ability to excite electricity.
The first use of the term "semiconductor" is of special interest. In 1792, (Ref. 127, p. 5) in repeating Galvani's frog leg experiments, Volta was sensitive to the loss of charge to the ground through various nominally nonconducting materials, such as ice, a damp wooden or marble table top, a person, or damp wall hangings, and called these "semiconducting." In 1805, (Ref. 186, p. 79) Ritter credits Volta as using the term "semiconductor" for materials such as the above. How-ever, further on (Ref. 186, pp. 225-239) in a more realistic discussion of semiconduction, Ritter states that Volta called such substances "semi-insulators", but that he prefers to call them "semiconductors." He did not have many examples, but mentions alcohol, ether, naphthene and metal oxides and their compounds.
The classification of conduction as "electrolytic" if electrode reactions are observed does not seem called for. Once the current has crossed the anode or cathode interface by means of an electron transfer reaction, the conduction through the medium is indistinguishable from metallic conduction, and Ohm's law is obeyed. This view, that there is fundamentally no difference between electrolytic and electronic conduction, has been expressed by a number of electrochemists, but their views have attracted little attention.
Thus, J.W. Richards187 reviewed earlier work by Marvin on induced currents in electrolytic conductors showing that they behaved in exactly the same manner as a metallic conductor of the same resistance. Richards concludes from this and his own experiments that solutions are simple conductors of the same nature as metallic conductors.
Kahlenberg188 reviewed the matter and favored the view that conduction in electrolytes is simple conduction the same as in metal, and that the reactions at electrodes are secondary phenomena.
Lukens189 made a laborious study of current distribution in metal and in solutions by means of equipotential lines, and concluded that the lines are identical in every respect in the solutions as in the case of the metallic conductor.
Brenner190 experimented with the induction of current in an electrolyte containing a dye, and without using electrodes, compared to electrolysis with electrodes. The difficulties of this type of experimentation are evident. Brenner concludes in favor of the migration of ions in the electrolyte, but this conclusion seems to represent his convictions more than clear cut results of the experiments.
Rosenberg and Postow (Ref. 191, pp. 161-190, an article on "Semiconduction in Proteins and Lipids") report electronic conduction in hemoglobin up to a hydration of about 20% and electrolysis and ionic conduction in the presence of more water.
The interpretation of these and other facts may be helped by placing the most important information available on the resistivity of different materials and media in a single chart for reference. This has been done in Fig. 31, which shows resistivities ranging from those of the best insulators such as polystyrene and mica to superconductors near the absolute zero of temperature. It is interesting that fused salts have such a narrow range of resistivity.
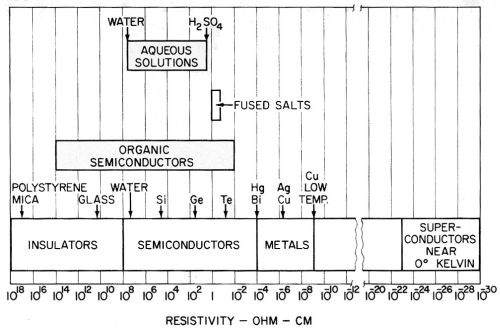
Figure 31 - Chart of the electrical resistivity of various media and materials at various temperatures.
This is one of the longest scales in existence for any single property. A defect is that it tends to disregard the effect of temperature variation. Thus, a metal may appear as a superconductor on the right hand end of the scale at zero degrees Kelvin, but move up to the middle of the scale into the range shown for metals, as the temperature increases to room temperature.
Semiconductors, on the other hand, move in the opposite direction as the temperature is increased, and exhibit a decreasing resistivity. The same is true of aqueous solutions with which we are largely concerned - the resistivity decreases as the solution is heated. From this point of view, solutions can be looked upon as liquid semiconductors, and there seems no reason for not considering them as such. Their resistivity is in the middle of the semiconductor range. The occurrence of barrier layers and electrolytic rectification at the electrodes is a commonplace. The occurrence of electrode reactions at the electrodes is perhaps evidence that this is the only means by which the current is able to cross the interface.
The study of "amorphous" or liquid semiconductors has developed into an extensive area of investigation. Gubanov192 does not discuss the problems of ionic conduction in liquid electrolytes, but does conclude that there is no basic difference between liquid and crystalline electronic conductors, and that the mechanism of electron motion in liquids and crystals is of the same nature.
The electronic properties of metal surfaces are of concern to solid state physicists. A noteworthy review is that of H.J. Juretschke (Ref. 90, pp. 38-53) who calls attention to a proposal of C. Herring193 of a fictitious or artificial metal called "jellium metal." Jellium metal might be considered to be a positive jelly or a uniform positively-charged medium against which the electrons move. These considerations are apparently helpful in discussing the nature of metal surfaces. The most recent review by Boudreaux and Juretschke194 is not available to the writer.
Bogenschütz (Ref. 195, pp. 319-368) gives an outstanding review of electronic devices based upon an electrochemical foundation, of which there are a surprising number. Some of this material also appeared earlier in several magazines.196 There are coulometers and microcoulometers, and electrochemical timers. Eleven pages are devoted to electrolytic information storage cells. Devices based on redox reactions include an electrolytic analog transistor, the Solion and a vibrating transducer. Conductivity cells appear in several devices. Electrolytic rectifiers and condensers are, of course, old and well-established pieces of equipment. Other devices mentioned are a gastrointestinal sender, an electrolytic respirometer, and a stabilization cell. Dry batteries, storage cells, fuel cells, and thermogalvanic cells are also briefly described.
It is, thus, apparent that the electronic industry already has close ties with electroplating and electrochemistry, and uses these freely in its device technology. It should be mentioned that the book by Vijh197 has also been helpful.












Related Content

Finishing High Reliability, Function Critical Parts
From safety critical automotive and aerospace components to lifesaving medical micro-components and implantable devices, Indiana-based Electro-Spec finishes applications that require zero failure rates.
Read MoreChecon Expands Market Offering with Acquisition of Umicore Electrical Materials USA, Inc.
The company is strategically working to meet future growth opportunities surrounding the use of precious metals and conductive materials like copper and aluminum.
Read MoreRead Next

Education Bringing Cleaning to Machining
Debuting new speakers and cleaning technology content during this half-day workshop co-located with IMTS 2024.
Read More
A ‘Clean’ Agenda Offers Unique Presentations in Chicago
The 2024 Parts Cleaning Conference, co-located with the International Manufacturing Technology Show, includes presentations by several speakers who are new to the conference and topics that have not been covered in past editions of this event.
Read More
Episode 45: An Interview with Chandler Mancuso, MacDermid Envio Solutions
Chandler Mancuso, technical director with MacDermid Envio discusses updating your wastewater treatment system and implementing materials recycling solutions to increase efficiencies, control costs and reduce environmental impact.
Read More