
The Characterization of the Stannous Chloride/ Palladium Chloride Catalysts for Electroless Plating
The paper is a 1975 study of just how the tin chloride / palladium chloride system worked for plated plastics, and which systems were most effective in doing the job.



by
E. Matijević,1 A.M. Poskanzer2 and P. Zuman1


1Institute of Colloid and Surface Science, Clarkson College of Technology, Potsdam, NY
2Shipley Co., Inc.. Newton, Mass.
The 1976 Carl E. Huessner Gold Medal Award for Best Paper appearing in Plating and Surface Finishing in 1975
Originally published as E. Matijević, A.M. Poskanzer and P. Zuman, Plating and Surface Finishing, 61 (10), 958-965 (1975).
Editor's Note: This paper is part of a series on the AES/AESF/NASF Best Paper Awards. In 1976, Dr. Egon Matijević and co-workers received the Carl E. Huessner Gold Medal Award for Best Paper appearing in Plating and Surface Finishing in 1975. A printable PDF version is available by clicking HERE.
ABSTRACT
Sixteen mixed PdCl2/SnCl2 catalyst compositions for electroless plating have been studied by electron microscopy, ultracentrifugation, polarography, light scattering and tested for catalytic functionality. A number of these were commercial samples, whereas the others were prepared as described in the patent literature. According to the authors, whose procedures were followed, some of the catalysts were claimed to be colloidal sols and some others were claimed to be true complex solutions. It is shown that all active catalyst compositions contained colloidal particles which could be separated by sufficiently long ultracentrifugation, and depicted by the electron microscope after direct deposition on grids. Functionality tests proved that all bottom layers separated by ultracentrifugation, containing the colloidal particles, were exceedingly efficient catalysts in electroless plating. None of the supernatant liquids, regardless of their coloration, showed catalytic activity.
One of the prerequisites for the electroless deposition of metals is the application of a catalyst to metallic (conductive) or non-metallic (dielectric) surfaces which are desired to be plated. The most commonly used catalyst systems consist of PdCl2 and SnCl2 in acidic (HCl) solutions. Originally, a two-step procedure was employed in which the substrate was first sensitized by immersion in an acidic tin (II) chloride solution followed by activation in a palladium chloride solution.1 Actually, Pearlstein2 showed that PdCl2 solutions alone applied at 125°F can catalyze a variety of substrates for electroless nickel deposition over the pH range 3.8-4.8, but a pre-dip in SnCl2 broadened the pH range (0.9-4.2) and allowed plating at room temperature.2 The two-step process had its deficiencies. For example, the copper clad substrate when immersed in a PdCl2 solution produced a flash coating of palladium metal which proved to be very expensive. Also, the flash coating was only weakly bonded to the substrate and, therefore, the bulk of it had to be removed by sanding or buffing to achieve a better bond in electroless plating.
A significant improvement in the catalytic process was the development of mixed PdCl2/SnCl2 catalytic systems which allowed the combination of the sensitization and the activation steps by immersion of the substrates in a single bath (one step catalyst). Essentially, all of these catalysts consist of dark liquids, containing high concentrations of HCl, in which various amounts of PdCl2 and SnCl2 were dissolved using different procedures. The first of these systems was described in a patent by Shipley3 in which it was claimed that the catalytic solution contains colloidal particles, which, upon adsorption on the substrates provide the catalytic sites for the reduction of metals, such as nickel or copper, to be plated out. Additional patents have been granted for PdCl2/SnCl2 catalyst systems which are supposed to contain exceedingly uniform colloidal particles of great stability.4
Interestingly, several other patents, aimed at improving Shipley's process, make the claim that the palladium/tin catalysts are true solutions and that their activity in electroless plating is due to the presence of complexes of various stoichiometric compositions.5,6
In view of the fact that all of the mentioned catalytic preparations3-6 contain essentially the same starting materials (PdCl2, excess SnCl2, HCl), and often differ little in the methods of preparation, a number of investigators have devoted a considerable amount of effort in order to decide whether the catalytic activity in electroless plating is due to colloidal particles or to some kind of Pd-Sn solute complexes. However, this has not resolved the "colloid" vs. “complex solutions” controversy, as advocates for either concept can be found. Thus, ultracentrifugation, electron microscopy and Mossbauer spectroscopy work by Cohen, et al.7-9 led these authors to conclude that the PdCl2/SnCl2 acid catalyst systems indeed are colloidal. A number of Japanese investigators arrived at the same conclusion.10,11
Contrary to this, Rantell and Holtzman claim that mixed PdCl2/SnCl2 catalysts are non-colloidal, and that the active components are solute complexes of the type SnPd7Cl16,12,13 although these findings have been challenged by Feldstein, et al.14 In their most recent papers, Rantell and Holtzman concede that commercial catalysts are indeed mixtures of solute complexes and colloids, but they maintain that the complexes are the more active components.15,16 The catalytic activity of soluble tin-palladium complexes has also been suggested by deMinjer and Boom.17
Thus the question as to whether the active catalytic component in the PdCl2/SnCl2 activator systems is colloidal or in the form of soluble complexes, or if it varies depending on the method of preparation, has not been resolved as yet. The ambiguity of the seemingly "simple" question as to whether a system is colloidal or non-colloidal* is caused by the fact that the catalysts under consideration are exceedingly dark colored solutions containing high concentrations of electrolytes. Furthermore, the techniques employed have not always been appropriate, and as such may have resulted in erroneous conclusions. For example, light scattering (more specifically the presence or the absence of a "Tyndall cone") has been commonly used as a test for the presence of colloidal particle. The appearance of an intense Tyndall cone certainly indicates the existence of colloidal particles, but colloidal systems may look optically clear, if there is no refractive index difference between the particles and the medium, and in this case a wrong conclusion may be drawn from light scattering effects.
Another common shortcoming of the cited studies is that most of them dealt with catalyst systems after their application to the substrate; a few investigations only concerned the catalyst liquid itself. By using appropriate techniques (e.g., ultracentrifugation), Cohen, et al. did establish the presence of colloidal particles in catalytically active solutions.7-9
Finally, in some instances the question of colloidal vs. non-colloidal nature of the catalyst has led to "compromise'' statements which have no scientific foundation. For example, D’Ottavio talks about a "semi-colloidal" part of the catalyst.4 Obviously the "semi-colloidal" state has no physical meaning, and its introduction only adds to the confusion.
This work has been carried out to establish whether there are any differences between various formulations of mixed PdCl2/SnCl2 catalyst systems for electroless plating, and whether these differences are related to their state of dispersion, i.e., colloidal vs. true solution. Furthermore, it was intended to determine whether the colloidal fraction (if such existed) is catalytically active, or if the true solution components play the role of sensitizers and activators. For this purpose a variety of catalysts was studied; some of these were commercial samples whereas others were prepared by procedures which should either give "colloidal" or "true solution" systems. Special care was taken that the methods employed did not cause any changes in the systems while these were analyzed, so that no artifacts were introduced.
Experimental
Catalyst formulation
Table 1 - Results of tests with various PdCl2/SnCl2 catalysts.
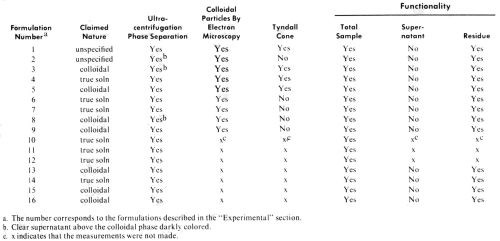
A total of 16 mixed PdCl2/SnCl2 catalyst systems has been studied, either commercially available, or prepared according to various procedures described in the patent literature. In the latter case instructions for the formulations were followed precisely as described in the corresponding patents or patent histories. These catalysts include systems claimed to be true solutions and others which are claimed to be colloidal dispersions. (See Table 1).
The formulations used were as follows (the same order is used in Table 1):
1. Example No. 1 of Japanese Patent 410824 (Peter Graham).
Palladium chloride (PdCl2) 1 g
Hydrochloric acid (HCl) (37% solution) 60 mL
Stannous chloride (SnCl2•2H2O) 22 g
Water to make 1000 mL
Palladium chloride was dissolved in one liter of water containing 60 mL of 37% hydrochloric acid. Dissolution was slow, and was aided by means of a magnetic stirrer. When the palladium chloride was completely dissolved, stannous chloride was added to the resulting solution.
2. Example No. 1 from the affidavit of Rudolph J. Zeblisky filed in U.S. Patent Application Serial 712,575 (Photocircuits Corp.).
Palladium chloride (PdCl2) 1 g
Hydrochloric acid (HCl) (37% solution) 300 mL
Stannous chloride SnCl2•2H2O 50 g
Water to 600 mL
Palladium chloride was dissolved in 300 mL of 37% hydrochloric acid. Stannous chloride was then added and dissolved with agitation by a magnetic stirrer. The solution was finally diluted with 600 mL of distilled water.
3. Example No. 2 from the affidavit of Rudolph J. Zeblisky, filed in U.S. Patent Application Serial 712,575 (Photocircuits Corp.).
Palladium chloride (PdCl2) 1 g
Hydrochloric acid (HCl) (37% solution) 300 mL
Stannous chloride SnCl2•2H2O 50 g
Water to 600 mL
Palladium chloride was dissolved in a solution prepared by mixing 600 mL of water with 300 mL of 37% hydrochloric acid. Stannous chloride (4 g) was added to the solution with vigorous agitation by a magnetic stirrer. The resulting solution was agitated for an additional 2 hr, and left to stand overnight. The remainder (46 g) of stannous chloride was then added slowly with agitation.
4. Example No. 3 from affidavit of Rudolph J. Zeblisky, filed in U.S. Patent Application Serial 712,575 (Photocircuits Corp.).
Palladium chloride (PdCl2) 0.25 g
Hydrochloric acid (HCl) (37% solution) 60 mL
Stannous chloride SnCl2•2H2O 12 g
Water to 1000 mL
Palladium chloride was dissolved in solution prepared by mixing 60 mL of 37% hydrochloric acid and 940 mL of distilled water. Stannous chloride was then rapidly dissolved in the above solution under vigorous agitation.
5. Example No. 1 from affidavit of Rudolph J. Zeblisky and Frederick W. Schneble, Jr., filed in U.S. Patent Application Serial 801,167 (Photocircuits Corp.).
Palladium chloride (PdCl2) 1 g
Hydrochloric acid (HCl) (37% solution) 300 mL
Stannous chloride SnCl2•2H2O 50 g
Water to 700 mL
Palladium chloride was dissolved in 300 mL of 37% hydrochloric acid and 700 mL of distilled water was then added. Stannous chloride (4 g) was dissolved in the resulting solution under vigorous stirring. The agitation was continued for 2 hr after which the solution was allowed to stand for 16 hr. Finally an additional 46 g of SnCl2•2H2O was added under vigorous agitation.
6. Example No. 2 from affidavit of Rudolph J. Zeblisky and Frederick W. Schneble, filed in U.S. Patent Application Serial 801,167 (Photocircuits Corp).
Palladium chloride (PdCl2) 4.05 g
Hydrochloric acid (HCl) (37% solution) 310 mL
Stannous chloride SnCl2•2H2O 202.5 g
Water to 397.5 mL
Palladium chloride was dissolved in a solution prepared by mixing 60 mL of 37% hydrochloric acid and 90 mL of distilled water. Stannous chloride was dissolved separately in a solution prepared by mixing 150 mL of 37% hydrochloric acid and 37.5 mL of water. The two solutions were combined and boiled for 1.5 hr (final volume ~360 mL). Finally, the solution was diluted with 100 mL of 37% hydrochloric acid and 270 mL of distilled water.
7. Example No. 1 of U.S. Pat 3,672,938 of Rudolph J. Zeblisky (Photocircuits Corp.).
Palladium chloride (PdCl2) 4 g
Hydrochloric acid (HCl) (37% solution) 70 mL
Stannous chloride SnCl2•2H2O 8.4 g
Water to 1000 mL
Palladium chloride and stannous chloride were placed into a flask and dissolved in 70 mL of 37% hydrochloric acid. Distilled water was then added to a total volume of 1000 mL. The solution was allowed to stand for approximately 1 hr at room temperature during which time the solution went through several color changes starting with blue-black, then turning dark green, deep brown and finally dark brown. At this time an additional 50 g of stannous chloride were added. The solution was then diluted with a solution of 60 g of stannous chloride per liter of 1.3N HCl to give a final composition of components:
Palladium chloride (PdCl2) 1 g/L
Hydrochloric acid (HCl) (37% solution) 100 mL/L
Stannous chloride SnCl2•2H2O 60 g/L
8. Example No. 2 of U.S. Pat. 3,011,920 of Charles R. Shipley, Jr., (Shipley Co.).
Palladium chloride (PdCl2) 1 g
Hydrochloric acid (HCl) (37% solution) 300 mL
Sodium stannate Na2SnO3•3H2O 1.5 g
Stannous chloride SnCl2•2H2O 37.5 g
Water 600 mL
Palladium chloride was dissolved in a solution prepared by mixing of 300 mL 37% hydrochloric acid and 600 mL distilled water. The sodium stannate was then added with vigorous agitation. This was followed by the addition of stannous chloride with vigorous agitation.
9. "Cuposit 6F" catalyst (Shipley Co.). Commercial sample.
10. Oxytron Activator 602 (Oxy Metal Corp.). Commercial sample.
11. Oxytron Activator 608 (Oxy Metal Corp.). Commercial sample.
12. Oxytron Activator 612 (Oxy Metal Corp.). Commercial sample.
13. Dynaplate Activator 120 (Dynachem Corp.). Commercial sample.
14. Enthone Activator 442 (Enthone Corp.). Commercial sample.
15. MacDermid D-34 (MacDermid Corp.). Commercial sample.
16. Cuposit 9F Catalyst (Shipley Co.). Commercial sample.
Of the above listed catalysts studied in this work, formulations 3, 5, 8, 9, 13, 15 and 16 were claimed to be colloidal sols, samples 4, 6, 7, 10, 11, 12 and 14 were claimed to be true solutions, and the nature of samples 1 and 2 was not specified (Table 1).
Experimental techniques
Special effort was made in this work to choose experimental techniques and to design experiments which permitted the necessary information to be obtained without causing a change of the catalyst system in the course of investigation.
Ultracentrifugation
In all cases, samples were subjected to ultracentrifugation at 25,000 rpm. Each formulation, prepared as previously described, was allowed to stand for 12 hr, and then 25 mL aliquots were ultracentrifuged for 9 hr. Polycarbonate tubes were employed in all experiments. A Beckman model L-2 preparative ultracentrifuge was used in this work with a type 30 rotor. In all cases, refrigeration of the rotor was utilized in order to keep the temperature at 20°C.
Immediately after removal from the ultracentrifuge, the samples were inspected visually for a phase separation, and then photographed to obtain a permanent record of the results. If a phase separation was observed, the residual fraction at the bottom of the ultracentrifuge tube was removed with a pipette and placed in a separate container. The supernatant fraction on top was removed to another vessel. Both the residue and supernatant fractions were saved for further testing.
For comparison purposes SnCl2 solutions were prepared in both 1N and 4N HCl at a concentration of 50 g SnCl2 per liter. These solutions were subjected to ultracentrifugation at 25,000 rpm for 24 hr after they stood for at least 12 hr. No phase separation could be detected in either 1N or 4N HCl solutions.
Electron microscopy
In order to use this technique properly, great care must be taken in preparing the samples for viewing in the electron microscope. The catalysts studied in this work contained high concentrations of dissolved salts. Therefore simple evaporation of a droplet onto an electron microscope grid, as done in some cases,12 is an unacceptable sample preparation technique, because the crystalline salts formed on evaporation would obscure the observations of the true composition of the system.
In the present work two different techniques were used. In the majority of cases specimens were prepared by dipping a 100 mesh copper grid coated with a collodion film into the catalyst liquid for about 1-2 min. The grid was then removed and excess liquid was gently blotted off, using Kimwipe tissue. This procedure prevents evaporation of the supernatant liquid on the grid and consequent contamination of the specimen by crystallized electrolytes.
In some instances, the electron microscope collodion coated grid was immersed in the catalyst system to be centrifuged and then the sample was spun. After extracting the grid from the liquid it was dried as described above. Both techniques carried out on the same sample showed the presence of identical particles, although the number of these was considerably larger when the collection was made by ultracentrifugation.
All electron microscope investigations were made in a Philips EM-100C instrument, which has a resolving power of ~15Å.
Light scattering
Angular scattering measurements were carried out in a Brice Phoenix light scattering photometer Series 2000 outfitted with Glan-Thomson prisms. A Brice Phoenix light scattering cell C101 was used in all experiments.
Each of the catalyst compositions studied was diluted with a stannous chloride-hydrochloric acid solution corresponding to the sample's acid and stannous ion concentrations. All final dilutions prepared for light scattering measurements contained 0.1 g PdCl2 per liter of catalyst liquid. The relative scattering intensities were determined for both horizontally and vertically polarized components of light at 10° intervals between the angles of 50° and 120°, using a mercury arc lamp without filters.
For purpose of comparison the light scattering of a "blank" solution containing 50 g of SnCl2•2H2O per liter of 4N HCl was also measured. Furthermore, the angular scattering data for a "monodispersed" polyvinyl chloride (PVC) latex sample with a modal particle radius of 0.17 μm and a number concentration of 1.1 × 106 particles per mL were also recorded under the same experimental conditions.
Polarography
Polarographic current-voltage curves were measured using a dropping mercury electrode (drop time t1 = 1.9 sec; outflow velocity, m = 2.3 mg/sec at height of the mercury column h = 65 cm) in a Kalousek cell with a saturated calomel electrode separated by liquid junction at 20°C, by means of the Princeton Applied Research polarograph Mark PAR 174.
The polarographic procedure has been employed for determination of ionic forms of palladium in the supernatant solutions obtained by ultracentrifugation of commercial or synthetic catalyst samples. Each formulation was aged for one week prior to ultracentrifugation, upon which the supernatant solutions were separated and a small portion was used for polarographic testing. For the analysis samples were diluted 10 times with either 4N or 1N HCl (to keep the HCl concentration identical with that in the catalyst). Parallel experiments were carried out with the same samples, but diluted 10 times with distilled water, giving the final concentrations of hydrochloric acid 0.4N and 0.1N, respectively.
To evaluate the contents of the ionic palladium in the samples, calibration curves were recorded for solutions containing varying amounts of palladium (added as PdCl2) in hydrochloric acid solutions of all four (4N, 1N, 0.4N and 0.1N) concentrations used. The stock solution contained 1 g of palladium (II) chloride in 4N HCl.
The diluted sample or the calibration solution (10 mL total volume) was transferred into the polarographic cell, the dropping mercury electrode was inserted, and the solution deaerated by a stream of nitrogen for 3 min. The zero current position of the pen was recorded, connection between the sample and reference compartments established by opening the central stopcock, and the curve was recorded from 0.0 V to ~0.6 V at a proper recorder sensitivity.
As palladium ions are reduced at positive potentials vs SCE, the current rises at zero applied voltage. Evaluation of the palladium ion concentration was carried out by measuring the difference between the limiting current at -0.1 to -0.3 V and the zero current line. At concentrations used, when recorder sensitivity was of the order of 0.5 μA/cm, capacity current was negligible and no correction was necessary.
Functionality testing
Testing for catalytic activity was carried out with all samples to decide if there were any differences between various formulations. More importantly, the functionality tests were performed on different fractions of the catalyst liquids after these had been subjected to ultracentrifugation. The latter was done specifically to determine if phases separated by ultracentrifugation exhibited different degrees of catalytic effects.
To determine the functionality of various catalyst formulations (or fractions thereof), epoxy laminate boards 2.5 × 10 cm (1 × 4 in.) were sensitized and plated in a copper plating bath using standard cleaning, sensitizing and plating procedures. Epoxy laminate boards were treated as follows:
- Immerse in boiling "Alconox" solution** 10-15 min to remove impurities.
- Rinse with water.
- Immerse in hot Al-chelate bath*** for 10-15 min to remove last traces of foreign organic matter from the board surface.
- Immerse in a HCl solution 15 vol%.
- Immerse in the catalyst formulation for 10 min.
- Rinse with water.
- Immerse in H2SO4 solution (5 vol%) for 1-5 sec.
- Rinse with water.
- Immerse in copper plating bath† for 5 min.
- Rinse with water.
Each catalyst was tested for functionality, without ultra-centrifugation, as prepared (at full strength) except for formulations No. 5 and 6 since these were intended to be concentrates as described in the original reference. Samples No. 5 and 6 were diluted as follows:
One mL of a solution containing 25 g of SnCl2•2H2O in one liter of dilute hydrochloric acid [1 part HCl (37%): 2 parts of water] was mixed with 250 mL of the hydrochloric acid of the same dilution. To this solution was added 0.875 mL of the prepared catalyst formulation to obtain dilution A. This procedure was repeated using 1.25 mL, 2.50 mL and 5.00 mL of the prepared catalyst formulation to obtain dilutions B, C, and D, respectively.
After each catalyst formulation was prepared, aged for about one week, and ultracentrifuged, the supernatant and residue fractions were separated as described previously. Both the supernatant and the residual fractions were then tested for catalytic functionality by the procedure given above. One mL of the supernatant and one mL of the residue was used for these tests. Due to the small volumes of sample available for this work, the epoxy-laminate boards could not be completely immersed in the sample for sensitization. Instead, the sensitizations were carried out by laying the boards flat on a beaker and spreading the sensitizing sample from a medicine dropper on the top side of the board. The evaporation of the residual phase or precipitation of the catalyst on the epoxy board was carefully avoided. After 10 min the board was rinsed with water and 5 vol% H2SO4. All other aspects of the functionality tests were identical to those described earlier.
Results
Ultracentrifugation
In all catalyst samples studied (Formulations 1-16), solid phase separation was observed upon ultracentrifugation (Table 1). Figures 1 and 2 show photographs of ultracentrifuge tubes before (left) and after (right) centrifugation of samples No. 9 and 10, respectively. Note (Table 1) that formulation No. 9 is claimed to yield a colloidal catalyst whereas Formulation 10 is claimed to be a true solution.
Sharp separation of boundaries between the bottom and top layers were observed in all samples at the end of the ultracentrifugation experiments. The supernatant liquids above the settled phase were in all systems optically clear, and in most cases colorless. Only three formulations (2, 3, and 8) yielded deeply colored (but perfectly clear) supernatant liquids after ultracentrifugation, indicating the presence of solute complexes. All bottom layers (residues) were exceedingly dark colored and opaque.

Figure 1 - Catalyst sample No. 9 (Table 1) before (left) and after (right) centrifugation at 25,000 rpm for 9 hr.
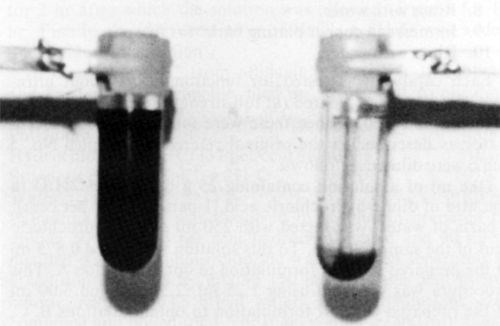
Figure 2 - Catalyst formulation No. 10 (Table 1) before (left) and after (right) centrifugation at 25,000 rpm for 9 hr.
Electron microscopy
Ten catalysts were investigated by electron microscopy as prepared before ultracentrifugation, and all of them showed the presence of colloidal particles. The latter varied in size and shape as illustrated in Figs. 3, 4 and 5 obtained with samples 6, 9, and 1 respectively. According to specification, sample 6 is supposed to be a true solution, sample 9 colloidal and no specification is given for sample 1. The particles in Figs. 4 and 5 are rather similar in shape and size, and obviously crystalline, whereas the particles shown in Fig. 3 are irregular and very likely amorphous. No difference in particle size and shape was observed in samples collected directly from the liquid catalyst on the electron microscope grid, or deposited by centrifugation, although in the latter case the number of particles was considerably larger. If the deposition by ultracentrifugation was carried out for too long, the particles completely coated the grid, making the electron microscopic investigation impossible.
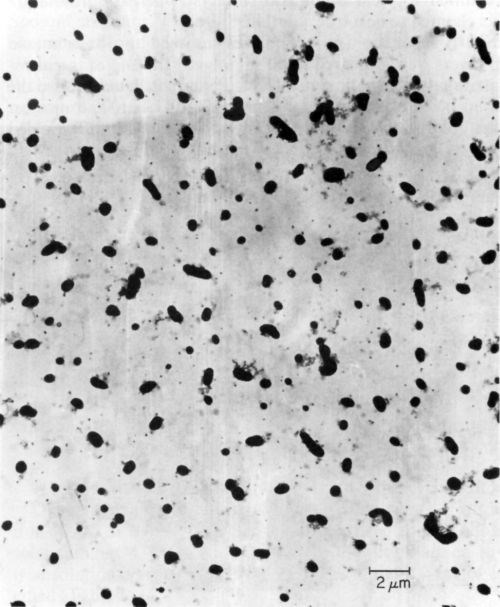
Figure 3 - Electron micrograph of colloidal particles in catalyst sample No. 6 (Table 1).
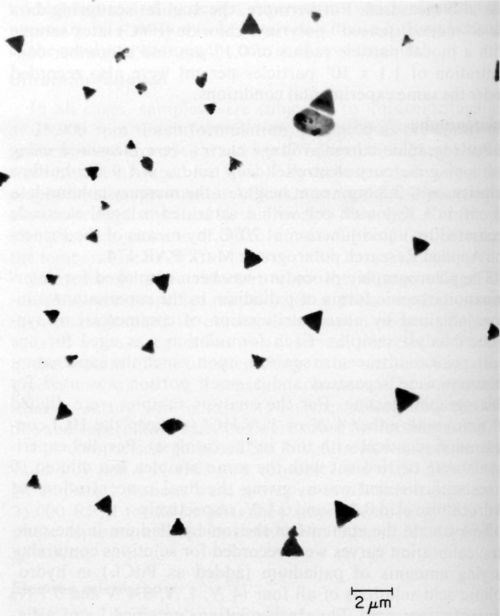
Figure 4 - Electron micrograph of colloidal particles in catalyst sample No. 9 (Table 1).
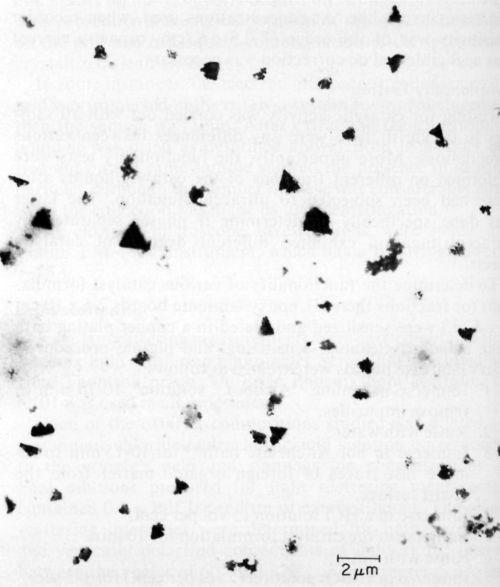
Figure 5 - Electron micrograph of colloidal particles in catalyst sample No. 1 (Table 1).
Two supernatant liquids after ultracentrifugation were also examined by electron microscopy. For this purpose, the colorless (very light yellow) supernatant from Formulation No. 1 and the deeply colored supernatant from Formulation No. 2 were used. Most of the grids showed no particles. On some grids a very few particles could be seen, which were, as a rule, considerably smaller than those separated in the bottom layer. Most likely the particles found in the supernatant were too small to settle on spinning, or diffused back during the separation of the two layers after ultracentrifugation.
Light scattering
Figure 6 contains the light scattering data for the blank (a stannous chloride solution), the PVC latex and Formulations No. 1 through 9, diluted as described in the experimental section. Relative scattering intensities for vertically polarized light are plotted as a function of the angle of observation. Negligible or no scattering intensity was observed with samples No. 2, 6, 7, 8 and 9, whereas catalysts No. 1, 3, 4 and 5 scattered light, although the angular dependence of the intensity varied from case to case.
Thus, despite the fact that all samples contained colloidal matter, some catalysts scattered light, whereas others did not. As a rule samples giving a deeply colored supernatant on ultracentrifugation showed no scattering. This could be explained in a number of ways. The highly absorptive supernatant liquid may have absorbed any scattered light so that it was not detected, or the colloidal particles may, in fact, have been sufficiently similar in refractive index to the supernatant liquids that no scattering would occur.
This shows that light scattering is not a valid test for the presence of colloidal particles in PdCl2/SnCl2 catalysts used for electroless plating. The absence of a Tyndall cone in these catalysts is not a sufficient criterion for establishing the nature of the catalyst system.
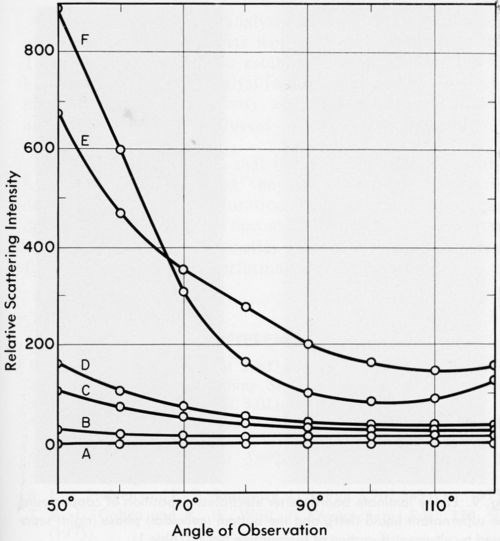
Figure 6 - The angular dependence of relative scattering intensities: Curves: A, Blank (50 g SnCl2•2H2O dissolved in 1 / 4N HCl) and formulations No. 2, 6, 7, 8 and 9: B, No. 1; C, No. 5; D, No. 3; E, No. 4; F, PVC latex, modal radius 0.17 μm, 1.1 × 106 particles/mL. All formulation numbers refer to Table 1.
Polarography
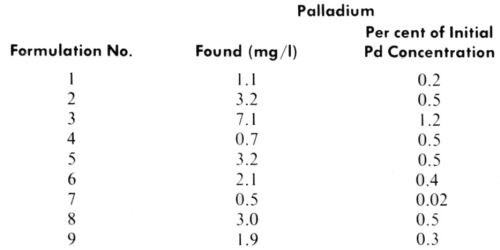
The limiting current observed at positive potentials corresponds to the concentration of palladium ions, both free and those which can be formed by dissociation of labile complexes (e.g., chloride complexes). Results, summarized in Table 2, indicate that only a very small fraction of the original palladium content exists in the supernatant solutions as free ions or bound in labile complexes. Dilution of the samples with distilled water rather than with hydrochloric acid solution had no effect on the analytical data. This indicates the absence of unstable complexes which would yield palladium ions at lower chloride concentrations, but does not exclude the possibility of changes in structure of the stable complexes or even formation of colloidal or solid particles from these stable complexes.
Table 2 - Polarographic determination of ionic palladium in supernatant solutions after centrifugation of catalyst liquids.
Functionality tests
Without centrifugation
All formulations prepared, as well as all commercial samples, were fully catalytically active. Formulations No. 5 and 6 were also functional after dilution as previously described.
With centrifugation
Striking differences in catalytic activity were observed between the residual layers and the supernatant liquids separated by ultracentrifugation. In all cases, the supernatant liquid showed no catalytic effects whereas all the residues were very highly functional.
It is essential to recognize that no functionality was observed with supernatant solutions regardless of their color, and thus of their chemical composition. On the contrary, all bottom layers containing the colloidal phase showed complete functionality, despite the difference in the particle size and shape detected by electron microscopy. Figures 7, 8 and 9 illustrate the results of functionality tests obtained with formulations 3, 5 and 6, respectively, after ultracentrifugation. In all these cases supernatant liquids showed no catalytic effects as no plating took place (left boards). The right hand side boards showed that complete plating occurred when the residue (the bottom layer after ultracentrifugation) was used as the catalyst. Of the three examples chosen, samples 3 and 5 were claimed to be colloidal and sample 6 true solution (Table 1). More importantly, the supernatant liquid used in the functionality test of sample 3 was darkly colored, indicating the presence of a certain amount of Pd-Sn solute complexes, yet it showed no catalytic activity. Indeed, sample 3 had the highest percentage of palladium remaining in the supernatant solution of all formulations tested (Table 2).
It is of further interest that a number of functionality tests were performed with freshly prepared, unaged catalyst Formulations 1-8. Samples 1, 4, 5, 6 and 8 showed only trace functionality when used 30 min after the catalyst systems were made up. Samples 2, 3 and 7 were only approximately 50% functional 30 min after their preparation.

Figure 7 - Epoxy laminate boards after electroless deposition of copper using the supernatant liquid (left), and the bottom (colloidal) phase (right) separated by ultracentrifugation of sample No. 3 (Table 2).

Figure 8 - Epoxy laminate boards after electroless deposition of copper using the supernatant liquid (left), and the bottom (colloidal) phase (right) separated by ultracentrifugation of sample No. 5 (Table 2).

Figure 9 - Epoxy laminate boards after electroless deposition of copper using the supernatant liquid (left), and the bottom (colloidal) phase (right) separated by ultracentrifugation of sample No. 6 (Table 2).
Discussion
There is no doubt that on mixing palladium chloride and stannous chloride solutions, acidified with HCl, solute complexes form at first. This is clearly indicated by the appearance of deep coloration on mixing of the two major solution components, which property has been used for quantitative assays of palladium by spectrophotometric techniques.18,19 Although the nature of the complex species has not been determined with certainty, it is generally agreed that they contain palladium bound to SnCl3- groups.20,21 The color of the solutions containing complexes depends on the method of preparation, and changes with time, indicating that more than one complex could be present. Such complexes might be in equilibrium which would make the isolation of individual species difficult.
On aging the PdCl2/SnCl2 catalyst solutions, colloidal particles eventually form; their size and shape varies according to the method of system preparation, concentrations of various components, temperature, etc. Attempts have been made to identify the composition of the particulate matter which precipitates in PdCl2/SnCl2 catalysts systems. Thus, it was suggested the solid to be metallic palladium obtained by reduction of Pd+2 by Sn+2 within the complex itself.11 Palladium and PdO were detected using electron diffraction on the catalyst formed by the two-step procedure.22 However, the formation of elemental palladium in significant quantities by reduction of Pd(II) with Sn(II) was doubted by Feldstein and Weiner.23 Instead, an amorphous alloy of the composition Pd3Sn was suggested.14
Cohen and West7 indicated that a complex Pd(II), 3Sn(II) is at first formed which is unstable and autoreductive, yielding a Sn-Pd alloy. Obviously, at present there is no unanimity on the nature of colloidal particles contained in PdCl2/SnCl2 catalysts.
The purpose of this work was not to identify the composition of the solute complexes or that of the colloidal particles. Rather, it was intended to establish if these catalysts are colloidal sols, and if the particles are the catalytically active component as claimed by some. Alternatively, it was endeavored to find how much, if any, of the palladium is left in the ionic form or as labile complexes and whether the latter are performing the role of the catalyst.
This work, which dealt with 16 different PdCl2/SnCl2 catalyst formulations for electroless plating, showed without doubt that all of them contained colloidal particles which could be separated by ultracentrifugation. Furthermore, all samples that were checked by electron microscopy confirmed the existence of particulate matter. Every precaution was taken to eliminate the possibility of precipitation during the sample preparation for electron microscopy. Needless to say, particles cannot form on ultracentrifugation; only existing particles can be separated. Thus, despite different claims made for the various catalyst systems studied, one must conclude that they are all colloidal sols.
Furthermore, it was conclusively established that on separation in the ultracentrifuge only the bottom layer containing the colloidal matter was catalytically active in all formulations. In no case did sensitization take place when the supernatant liquid was used from which the colloidal particles had been removed. It is of particular importance that even the darkly colored (but optically clear) supernatants showed no catalytic activity. As indicated by the color, these solutions must have contained small amounts of Pd-Sn solute complexes.
Polarographic data show that practically no ionic palladium or its labile complexes are left in the supernatant liquids. Thus, it is fair to conclude that the mixed PdCl2/SnCl2 catalysts for electroless plating owe their activity to the presence of colloidal particles which contain most of the palladium. The latter case particularly applies to formulations which gave colorless or weakly colored supernatant liquids after centrifugation. Conversely, no catalytic activity could be observed when catalyst liquids were freed from the colloidal particles, regardless of the coloration of these solutions; i.e., the complex solutes did not act as catalysts.
Systematic chemical analysis of the colloidal particles present in various catalysts would now be highly desirable. This would enable one to establish the mechanism and the kinetics of the colloid catalyst formation as well as the mechanisms of the catalytic activity, and would greatly advance the understanding of the process of electroless deposition of metals.
It is of further interest that immediately after the catalyst formulations are prepared, they are of limited functionality despite their intensive coloration. Only after prolonged aging do the same formulations become fully functional. This would again indicate that the kinetics of colloid formation plays a fundamental role in the performance of the catalysts.
References
1. E.A. Bergstrom, U.S. Patent 2,702,253 (Nov. 1950).
2. F. Pearlstein, Metal Finishing, 53, (8), 59 (1955).
3. C.R. Shipley Jr., U.S. Patent 3,011,920 (Dec. 1961).
4. E.D. D'Ottavio, U.S. Patent 3,532,518 (Oct. 1970); U.S. Patent 3,650,913 (Mar. 1972).
5. R.J. Zeblisky, U.S. Patent 3,672,938 (June 1972); U.S. Patent 3,682,671 (Aug. 1972).
6. E.J. Fadgen and E.B. Saubestre, U.S. Patent 3,767,583 (Oct. 1973).
7. R.L. Cohen and K.W. West, J. Electrochem. Soc., 120, 502 (1973).
8. R.L. Cohen and K.W. West, Chem. Phys. Letters, 16, 128 (1972).
9. R.L. Cohen, J.F. D'Amico and K.W. West, J. Electrochem. Soc., 118, 2042 (1971).
10. M. Kose, T. Kishi, H. Yamamoto and T. Nagai, Kinzokuhyomen-gijutsu (J. Metal Finishing Soc, Japan), 24, 203 (1973).
11. M. Tsukahara, T. Kishi, H. Yamamoto and T. Nagai, ibid., 23, 83 (1972).
12. A. Rantell and A. Holtzman, Plating and Surface Finishing, 61, 326 (1974).
13. A. Rantell and A. Holtzman, Trans. Inst. Metal Finishing, 51, 62 (1973).
14. N. Feldstein, M. Schlesinger, N.E. Hedgecock and S.L. Chow, J. Electrochem. Soc., 121, 738 (1974).
15. A. Rantell and A. Holtzman, Trans. Inst. Metal Finishing, 52, 31 (1974).
16. A. Rantell and A. Holtzman, Electroplating and Metal Finishing, 27 (2), 15 (1974).
17. C.H. de Minjer and P.F. J. v.d. Boom, J. Electrochem. Soc., 120, 1644 (1973).
18. G.H. Ayres and J.H. Alsop III, Anal. Chem., 31, 1135 (1959).
19. W.B. Pollard, Analyst, 67, 184 (1942).
20. G.E. Batley and J.C. Bailar, Inorg. Nucl. Chem. Letters, 4, 577 (1968).
21. M.A. Khattak and R.J. Magee, Chem. Commun., 17, 400 (1965).
22. R. Sard, J. Electrochem. Soc., 117, 864 (1970).
23. N. Feldstein and J.A. Weiner, J. Electrochem. Soc., 120, 475 (1973).
Footnotes:
*For a comprehensive discussion of the definition of colloidal state and of the scope of colloid science, see: E. Matijević, Chemical Technology, 3, 656 (1973).
**Trade name of Shipley Company.
***An alkaline cleaner (Shipley Company).
†Shipley Company “Copper Mix 328” proprietary electroless copper plating bath.
About the authors


















Related Content

Conveyors and Paint Systems
Choosing the right conveyor system, coating technology, and ancillary equipment.
Read More
Anodizing for Bonding Applications in Aerospace
Anodizing for pre-prep bonding bridges the gap between metallic and composite worlds, as it provides a superior surface in many applications on aluminum components for bonding to these composites.
Read More
Zinc Phosphate: Questions and Answers
Our experts share specific questions about zinc phosphate and pretreatment
Read More
How to Maximize Nickel Plating Performance
The advantages of boric acid-free nickel plating include allowing manufacturers who utilize nickel plating to keep up the ever-changing regulatory policies and support sustainability efforts.
Read MoreRead Next

Delivering Increased Benefits to Greenhouse Films
Baystar's Borstar technology is helping customers deliver better, more reliable production methods to greenhouse agriculture.
Read More
A ‘Clean’ Agenda Offers Unique Presentations in Chicago
The 2024 Parts Cleaning Conference, co-located with the International Manufacturing Technology Show, includes presentations by several speakers who are new to the conference and topics that have not been covered in past editions of this event.
Read More
Episode 45: An Interview with Chandler Mancuso, MacDermid Envio Solutions
Chandler Mancuso, technical director with MacDermid Envio discusses updating your wastewater treatment system and implementing materials recycling solutions to increase efficiencies, control costs and reduce environmental impact.
Read More